
A digital health startup wraps up a strong meeting with a pharma sponsor’s medical affairs team. The pitch lands and next steps get scheduled. A week later, the sponsor’s regulatory team sends over a tech requirements document. Half the terms on the page are new to the CTO: combination product, 21 CFR Part 11, GxP, validated software, data use agreement, co-promotion compliance. These are the table stakes for building a companion app: pharmaceutical partnership requirements that change the shape of the deal.
A pharmaceutical company partnership built around a digital companion app is a regulated product relationship. Treat it like a distribution deal with compliance overhead and you’re already losing the negotiation. The FDA framework attached to the drug becomes the design brief for the app:
- PhRMA-aligned promotional rules,
- 21 CFR Part 11 records discipline,
- and device controls if it’s a combination product.
Pharma spends more than $5 billion annually on patient support programs in the US, and digital companion apps are the primary vehicle for that spend. The gap between what digital health companies expect to build and what pharma compliance teams require is the most common reason these partnerships stall after term sheet
What does building a companion app for a pharmaceutical partnership require?
A pharma companion app requires combination product classification, HIPAA infrastructure with SOC 2 Type II, 21 CFR Part 11 audit trail where records reach FDA, IEC 62304 development process, configurable CMS for labeling compliance, and three-stack hub integration via Surescripts: NCPDP SCRIPT for ePrescribing, FHIR Specialty Patient Enrollment, and proprietary REST APIs per hub.
Key takeaways
- Compliance requirements flow from the drug’s regulatory status to the app’s architecture. They’re real, specific, and non-negotiable. A pharma technology partnership is a regulated product relationship from the first term sheet onward.
- The requirements are discoverable before signing. Combination product status, REMS implications, 21 CFR Part 11 applicability, IP and data ownership: all knowable with the right questions at the right time.
- The deals that close go to teams who understand the regulated stack before negotiation. What you understand about the stack determines what you can negotiate and what timeline you can credibly commit to.
Table of contents
- The first question pharma’s regulatory team will ask: is this a combination product?
- REMS programs add a compliance layer HIPAA doesn’t cover
- The technical due diligence checklist: what pharma’s team will audit before signing
- Co-promotion compliance: when the app is marketed alongside the drug
- What to build differently: the pharma-ready technical architecture
- The realistic timeline: from term sheet to pharma-deployable product
- Why choose Topflight for pharma companion app development
The first question pharma’s regulatory team will ask: is this a combination product?
Most drug companion apps are. A combination-product digital health app sits under 21 CFR Part 3 and gets reviewed by the FDA Office of Combination Products (OCP), and that classification governs which compliance stack applies and how long the build runs. Pharma’s regulatory affairs team makes this call before engineering ever sees the term sheet.
A combination product is any product that pairs a drug (or biologic) with a device under one regulated use. Your app becomes a constituent part. Both frameworks apply at the same time, with one center inside FDA (CDER, CBER, or CDRH) assigned as the lead based on primary mode of action (PMOA).
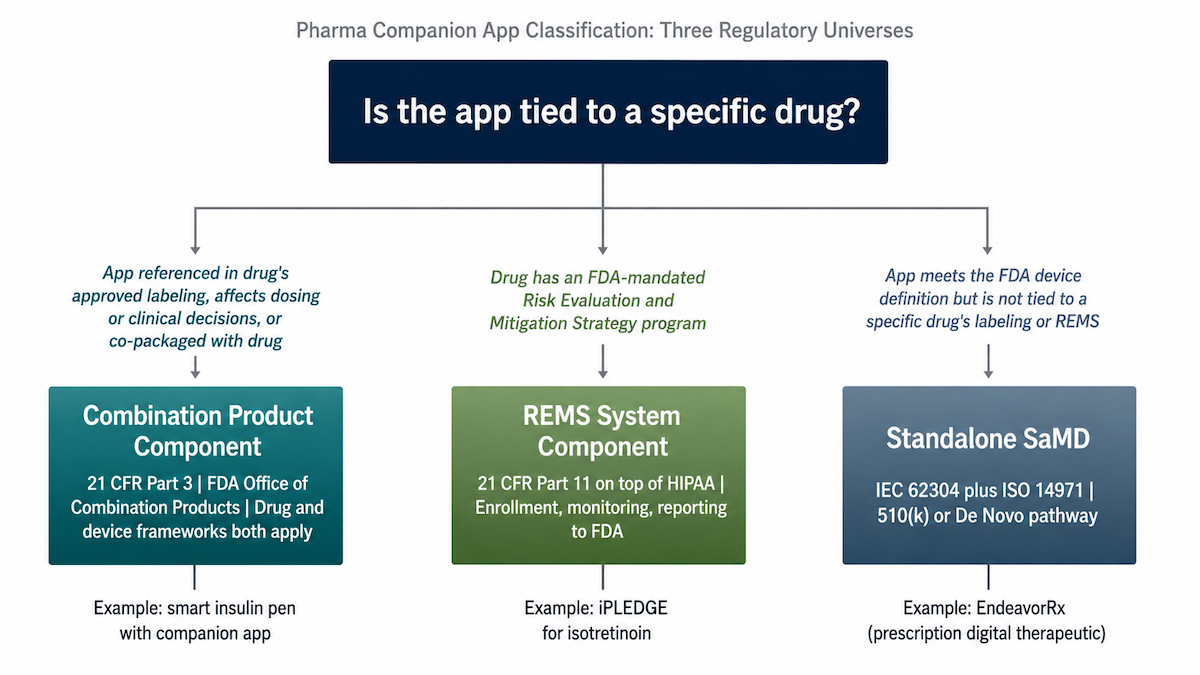
Most drug companion apps hit two of these three factors
- The app is referenced in the drug’s approved labeling.
If the NDA or BLA names the app as part of the prescribed use, the app is part of the regulated product. That’s the cleanest path to combination product status.
- The app affects dosing, administration, or clinical decision-making.
Titration support or injection guidance, anything that changes how the drug actually gets used.
- The app is co-packaged with the drug.
A co-packaged combination product is the textbook case: shared box, shared dispensing channel.
One factor moves you into accessory classification territory; two or three lands you fully inside combination product status. The OCP makes the formal call through a Request for Designation.
A standalone prescription digital therapeutic (PDT) without a paired drug is a different animal: it’s a digital therapeutic (DTx) regulated as a device, with no drug-digital combination on the file. Co-promotion alone doesn’t reclassify it. Promotional rules apply, but the combination product framework doesn’t activate.
The 2026 FDA deregulatory wave doesn’t reach you
There’s a trap in this year’s headlines. The January 6, 2026 FDA guidance updates reduced oversight on a defined slice of standalone digital health: certain clinical decision support tools and low-risk consumer wearables. The agency also withdrew the SaMD International Clinical Evaluation guidance the same week.
If you’ve read the coverage and assumed the regulatory bar dropped across the board, the scope is worth a second look. None of these changes apply when a drug is in the picture. The 2026 wave is about standalone software. A drug companion app sits inside the combination product framework, and that framework didn’t move.
Translation: your stack still carries combination product weight. Part 11 still applies where records flow to FDA, and device controls still apply on the device side.
Before signing a development contract, run a regulatory affairs assessment of combination product status. Regulatory affairs owns this call, and answering it late is the most expensive mistake in the project. For broader context, see our work on medical device companion app development and our explainer on whether your health AI is a medical device.

REMS programs add a compliance layer HIPAA doesn’t cover
If the drug carries a REMS program, your companion app needs to satisfy 21 CFR Part 11 on top of HIPAA. Of all the pharma companion app regulatory requirements we audit, this is the single most frequently missed.
REMS = Risk Evaluation and Mitigation Strategies. FDA-mandated safety programs for drugs with serious risks: clozapine, isotretinoin, mifepristone, opioid REMS, certain biologics.
When your app supports REMS compliance for the pharma sponsor (enrollment and consent capture, monitoring confirmations), the app inherits a chunk of the REMS regulatory perimeter.
REMS adds three requirements the app has to own
- Patient enrollment with a defensible audit trail, plus electronic consent. Both the enrollment record and the e-consent must hold up if they’re used in FDA REMS compliance reporting. That means 21 CFR Part 11 compliant electronic signature.
- Monitoring protocol compliance tracking with reportable output. Whatever the REMS imposes (lab monitoring, prescriber attestations) gets captured and surfaced to the pharma’s REMS program administrator in a format they can actually report.
- Part 11 compliance for any electronic record submitted to FDA in REMS reporting. Immutable, timestamped, user-attributed records with end-to-end data integrity.
NCPDP SCRIPT v2023011 carries REMS-specific transactions at the prescribing layer if your stack touches ePrescribing for a REMS drug.
Why Part 11 is the separate framework you can’t skip
Most digital health compliance programs we audit address HIPAA. They have BAAs (Business Associate Agreement), encryption posture, access controls, breach response. None of that satisfies 21 CFR Part 11.
HIPAA governs how you handle protected health information. Part 11 governs how electronic records and signatures behave when they’re submitted to FDA. Different rule, different audit. Conflating the two is how REMS app builds fail at audit, usually around month nine when the pharma compliance team sits down with the records.
If you’re building an adherence layer into a REMS app, our deep dive on clinical medication adherence platform development covers the data architecture side that connects here.
The technical due diligence checklist: what pharma’s team will audit before signing
Pharma’s tech and regulatory team audits the digital partner against a five-domain checklist before any term sheet. Building an app for pharma partnership? FDA requirements mean running the checklist against your own stack first.
Every line surfaces in pharma partner due diligence; better you surface it first.

Regulatory and classification
- Software safety class assigned with documented ISO 14971:2019 risk analysis
- FDA classification determined (SaMD companion app, accessory, or combination product component)
- IEC 62304:2006 + Amendment 1:2015 status documented: development plan, SOUP list, traceability matrix current
- If SaMD, FDA pre-submission meeting scheduled or completed with the clearance pathway identified (510(k) clearance, FDA De Novo, or investigational device exemption (IDE) where applicable)
Classification comes first; everything else flows from it. Routes: IEC 62304 compliance for startups, health AI FDA clearance.
Data security and privacy
- HIPAA compliant infrastructure operational: BAA chain documented, encryption at rest and in transit, audit logging
- SOC 2 Type II report available, or Type I with a 12-month roadmap to Type II
- Penetration testing within the last 12 months, findings remediated
- Data residency and localization confirmed (GDPR for EU drugs)
- Data use agreement (DUA) template available
- SDK and tracker audit completed across the mobile and web stack, with API security review
That last line traces back to GoodRx’s February 2023 FTC/DOJ settlement: $1.5M plus a 20-year compliance monitoring order for leaking user health data via embedded SDKs (Meta Pixel, Google Analytics, Criteo). Pharma security teams audit your SDK stack as a line item now. Run yours first. Background: HIPAA compliant software development.
Electronic records (if applicable)
- 21 CFR Part 11 assessment completed if app-generated records will reach FDA submissions or REMS reporting
- Audit trail architecture documented: append-only, timestamped, user-attributed, tamper-evident
- Electronic signature implementation confirmed where required
“If applicable” carries weight: many companion apps don’t trigger Part 11 at all.
IP and data ownership
- IP position documented and negotiable: what the builder owns, what is licensed to pharma under a technology licensing or co-development agreement, what is jointly developed (IP assignment terms explicit)
- Data ownership documented for PHI, de-identified aggregate, and real-world evidence (RWE)
- Exclusivity clause scope documented: is the platform exclusive to this pharma partner, this therapy area, or available to others
- De-identified data rights and data sharing agreement terms negotiated
Cleanest public reference: the 2019 Akili-Shionogi deal. Shionogi got Japan/Taiwan exclusivity only ($20M upfront, up to $105M in milestones). Akili kept global rights outside the territory plus platform IP and ex-territory data. Both sides defensible.
Clinical validation
- Usability testing completed with representative users; FDA requires human factors studies for many device categories
- Clinical evidence strategy defined: published study, active pilot, or registry plan
- If the app feeds clinical trial data into a study, GCP (Good Clinical Practice) applies, with software validation under Computer System Validation (CSV) and eClinical electronic data capture (EDC) integration on the build side
- IRB status documented if any data collection constitutes human subjects research, with GxP (Good Practice regulations) applicability mapped where relevant
Builder alert: data ownership and exclusivity are the two clauses you cannot under-negotiate
These two carry the longest tail. A pharma partner owning the app’s RWE acquires your most valuable long-term asset: the data behind outcomes-based contract negotiation, market access digital health positioning, and specialty pharmacy pull-through evidence.
Once signed away, no data sharing agreement claws it back. Exclusivity: locking the platform to one partner across therapy areas turns a Series B story into a single-customer dependency.
Negotiate data ownership and de-identified data rights in writing before the term sheet.
Co-promotion compliance: when the app is marketed alongside the drug
The moment the pharma partner co-markets your app with the drug, the rules change. Engineering decisions become promotional material decisions, and the drug’s approved labeling becomes the constraint on what the app can show. Digital therapeutic pharma partnership compliance starts here, well before launch.
Co-promotion changes the build in three practical ways.
PhRMA code governs your app’s HCP-facing content
The PhRMA Code on Interactions with Healthcare Professionals (current revision effective January 1, 2022) treats your companion app as a promotional asset once it’s presented to HCPs alongside the drug. That covers branded digital health surfaces in particular: onboarding screens that name the drug, clinical outcome claims, dosing references, data visualizations on the prescriber view.
All of it has to present balanced risk information consistent with the drug’s approved labeling. Same standard as every other piece of pharma promotional material, whether it’s a detail aid, an unbranded disease state site, or a sales rep leave-behind. The app doesn’t get a different bar because it’s software.
MLR review becomes part of your release cycle
Anything that references both the app and the drug gets reviewed by pharma medical affairs, legal, and regulatory teams before release. App store listings. In-app onboarding content. Digital marketing campaigns the pharma launches at HCP audiences. Non-branded digital health surfaces that bridge to the branded experience.
MLR cycles run weeks for routine assets, longer for anything novel. Build that timeline into your release cadence from sprint one or every content change becomes a launch delay.
Off-label risk lives in your data visualizations
This is the one engineering teams miss most often. If the app shows a clinical outcome that goes beyond the drug’s approved indications, even by implication through a trend line or an outcome tracker, that can constitute off-label promotion. The engineering team has to read the drug’s approved labeling before designing any clinical outcome tracking or data presentation feature.
The approved labeling defines what the app can claim. The pharma marketing team’s preferences and the product manager’s roadmap both sit downstream of that. Labeling first, design second.
What to build differently: the pharma-ready technical architecture
Building a pharma companion app diverges from a consumer healthtech app in five specific places, all architectural, all in sprint one. Each is cheap to design in from the start and expensive to retrofit when pharma’s compliance review opens six months later. Together they define the companion digital therapeutic regulated stack.

Immutable audit log architecture from day one
Any app generating data that ends up in pharma reporting needs an append-only event log at the database schema level. Each event timestamped and user-attributed, with tamper-evidence enforced at the schema level.
Modifiable audit logs are logging tables; Part 11 needs append-only event logs. Retrofitting append-only behavior into a mutable record structure is a major engineering project that lands mid-audit. Build it from day one.
Configurable CMS for labeling changes
Patient education and dosing guidance belong in a CMS the pharma regulatory team can update without an app store release. Same for safety messaging tied to label revisions. Drug label changes require the app’s content to stay consistent with the current label.
Hardcoded strings turn every label change into a full app store release plus an Apple and Google re-review, plus a deployment window where the app ships with stale labeling.
If any of the content is ML-driven, the December 2024 FDA PCCP guidance for AI-enabled device software functions lets the pharma team pre-specify model updates without a new submission. Worth designing in. Background on the broader picture: AI in healthcare compliance.
PHI and de-identified data separation at the schema level
The pharma partner needs de-identified aggregate data for:
- market access
- real-world evidence (RWE)
- real-world data (RWD) work
Identifiable PHI stays with the digital health company under HIPAA, with no authorization to share absent explicit patient consent.
Architect the separation at the schema level. Different databases, with separate access controls and audit trails. De-identification happens at write time. Filtering identifiers out of a query result on the way out is how PHI leaks into pharma data lakes.
Multi-tenant architecture, even for a one-drug build
If the strategy involves more than one pharma partner across different drugs, therapy areas, or geographies, single-tenant architecture creates a refactoring bill that comes due when partner two signs.
Design for separate data environments per drug or program. Configurable feature sets and independent audit trails sit at the partnership level. Even for a one-drug build, the marginal cost is small at the beginning and large later. HIPAA compliant app development covers the infrastructure underneath.
Hub integration runs across three distinct stacks
Hub integration is fragmented across three distinct stacks. A companion app touching pharma patient support program (PSP) hub services rides all three, with no single integration layer and no hub-published FHIR endpoint.
- NCPDP SCRIPT via Surescripts for ePrescribing and ePA. NCPDP SCRIPT dominates the script layer. Build to v2023011, the CMS-named version under the ONC HTI-4 Final Rule (certification deadline December 31, 2027; v2017071 sunsets January 1, 2028). Surescripts is your switch; building direct NCPDP connectivity takes years and rarely pencils out for a digital health team.
- HL7/NCPDP FHIR Specialty Medication Enrollment IG (R4), mediated by Surescripts. The only standardized FHIR integration path for moving enrollment data from EHR to hub. AssistRx, CareMetx, and PharmaCord all consume specialty enrollment through Surescripts Specialty Patient Enrollment. No hub publishes its own FHIR endpoint or CapabilityStatement.
- Proprietary REST APIs per hub for case status, dispensing confirmation, copay, and adherence. AssistRx Advanced Gateway, CareMetx Intellicore, and ConnectiveRx expose proprietary REST contracts. Expect a BAA and per-brand configuration; documentation arrives only after NDA. Budget eight to sixteen weeks per hub, more if the stack also touches Lash Group, Cardinal Sonexus, or Eversana.
EHR-side surfacing happens through vendor marketplaces: Veradigm Developer Network and AccelRx, Epic Connection Hub, athenahealth Marketplace. Direct SMART-on-FHIR launch from a hub is rare.
The one exception worth knowing is CoverMyMeds (McKesson), which ships a direct Epic MyChart SMART-on-FHIR app; its ePA still rides NCPDP SCRIPT, but retrieval uses Epic’s standard FHIR APIs.
As TrialCard put it in Pharmaceutical Commerce, “if you’ve seen one hub, you’ve seen one hub.” The standards exist; the implementations don’t converge.
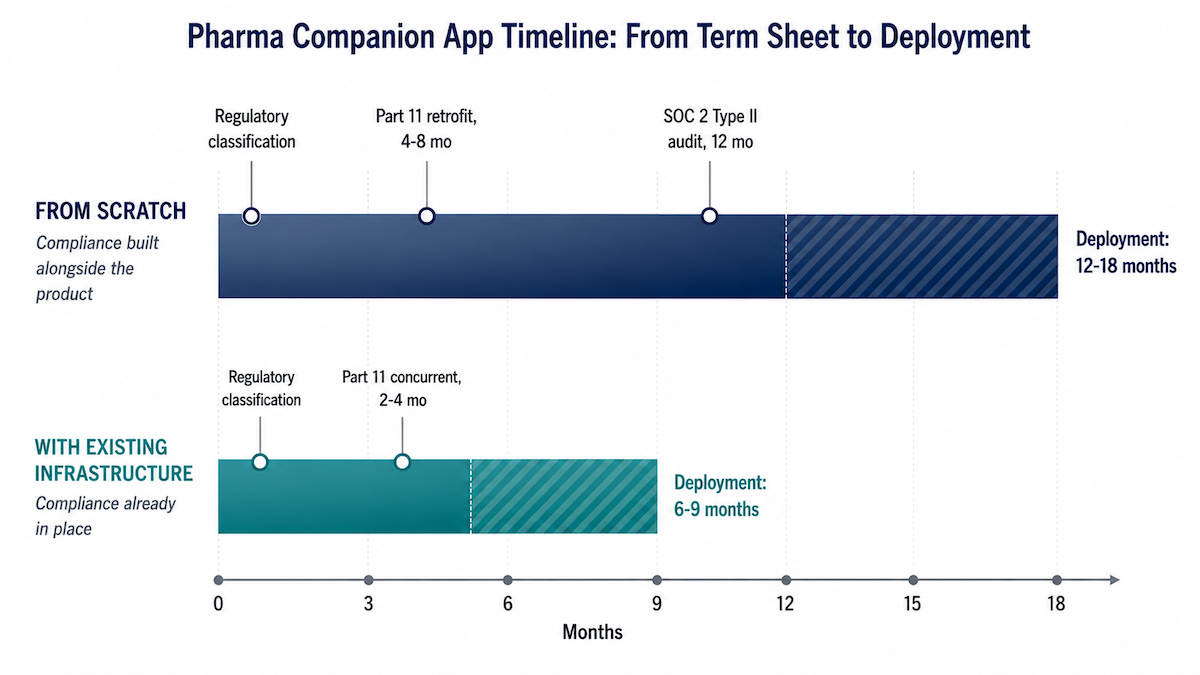
The realistic timeline: from term sheet to pharma-deployable product
Pharma timeline conversations go sideways when the digital health team quotes a build estimate without compliance overhead. Compliance milestones set the clock, and they run on their own schedule.
The compliance milestones that set the clock
In practice, the timeline usually bends around five milestones:
- Regulatory classification assessment: 4 to 6 weeks before development starts. Combination product vs SaMD vs accessory vs general wellness gets settled here.
- Pharma-ready MVP: 6 to 9 months depending on feature scope. A focused SaMD pharma companion app development build sits at the lower end; broader feature sets at the higher end.
- SOC 2 Type II: 12 months from audit start. Type I in 3 to 6 months if needed for the initial partnership, with a documented Type II roadmap.
- 21 CFR Part 11 assessment and implementation: 2 to 4 months concurrent with development when planned from the start, 4 to 8 months as a retrofit.
- FDA pre-submission meeting: 60 to 90 days from submission request to the meeting itself, plus prep time on your side.
Total realistic timeline: 12-18 months from scratch, 6-9 with existing infrastructure
From partnership term sheet to pharma-deployable product: 12 to 18 months for a digital health company building HIPAA, SOC 2, and IEC 62304 infrastructure alongside the product, 6 to 9 months for companies that already have all three in place.
The gap is where the conversation gets difficult. Companies with the infrastructure already built are carrying a different scope of work than companies assembling it from scratch alongside the product.
Why pharma’s default expectation is off
Pharma partners often arrive expecting shorter timelines because they’re comparing to software consulting engagements that skip compliance infrastructure entirely. Those builds don’t carry SOC 2 Type II prep, Part 11 architecture, or IEC 62304 design controls. The digital health company that can articulate that distinction wins the credibility conversation; quoting a longer number without the breakdown costs you the deal.
Topflight’s companion app development practice has run that conversation with sponsors many times, and the healthcare app developers on our team build to those milestones as baseline.
Why choose Topflight Apps for pharma companion app development
Topflight builds pharma-ready companion apps with the regulatory and technical infrastructure pharma partners actually audit for:
- IEC 62304-compliant development process (light-therapy app case study)
- HIPAA-compliant architecture
- 21 CFR Part 11 audit trail design
- configurable content architecture for labeling compliance
- usability validation for FDA submissions
Years of work on HIPAA-bound healthcare apps mean those pieces ship as one stack, ready for pharma audit on day one.
The strongest conversations happen when the digital health team can answer pharma’s compliance questions before they’re asked. That’s the work Topflight has done across companion apps, clinical platforms, and regulated digital therapeutics.
Landing a pharma partnership starts with understanding what their regulatory team will require of your stack. The digital health pharma partnership technology you bring to that conversation determines what you can negotiate and what timeline you can credibly commit to.
Talk to Topflight before the due diligence conversation starts.
FAQ
Does a companion app for a pharmaceutical product need FDA clearance?
It depends on classification. SaMD may need 510(k) or De Novo clearance based on risk class. Combination product components are subject to drug and device frameworks. General wellness software is not regulated as a device.
What is a combination product and does my pharma companion app qualify as one?
A combination product combines drug and device components under FDA regulation, subject to both frameworks. A companion app qualifies if it’s referenced in approved labeling, affects dosing or clinical decisions, or ships with the drug.
What is 21 CFR Part 11 and when does it apply to a companion app?
Part 11 is FDA’s framework for electronic records and signatures submitted to the agency. It applies when an app’s records reach FDA submissions or REMS reporting. Records must be immutable, timestamped, user-attributed, and tamper-evident.
What is a REMS program and what does it require from a companion app?
REMS = Risk Evaluation and Mitigation Strategy, FDA’s safety program for drugs with serious risks. The app must capture enrollment with electronic consent, track monitoring compliance, and meet Part 11 if records reach FDA.
What data ownership structure is typical in a pharma technology partnership?
Structures vary. The typical pattern: PHI stays with the app under HIPAA, de-identified aggregate data is pharma-owned or jointly licensed, platform IP stays with the digital partner. Defaults rarely favor the digital partner; negotiate explicitly.
What is the difference between a branded and non-branded pharma companion app?
Branded apps name the drug and trigger PhRMA Code requirements plus MLR review for content. Non-branded apps focus on the disease without naming the drug, carry fewer promotional constraints, and offer less co-marketing reach.
What does SOC 2 Type II require and do I need it for a pharma partnership?
SOC 2 Type II is an audit of security controls operating effectively over 12 months. Pharma partners typically require Type II, or Type I with a Type II roadmap. Plan 12 months from audit start.
How long does it take to build a pharma-ready companion app?
12 to 18 months from scratch for a digital health company building HIPAA, SOC 2, and IEC 62304 infrastructure alongside the product. 6 to 9 months for companies with that infrastructure already in place.