
Reviewed & updated July 2026
Your device is cleared. Or close. And the app, the thing patients actually touch every day, is either a known weak point or not yet built. Medical device companion app development is the conversation Topflight Apps has with device companies every week. The scale explains why.
As of 2024, 589 million adults live with diabetes, about 1 in 9, per the International Diabetes Federation’s Diabetes Atlas. WHO counted 363 million people with asthma in 2023. COPD affects more than 200 million and is the third-leading cause of death worldwide. CDC puts the US population living with a heart rhythm disorder at roughly 11 million.
Every one of those patients is a candidate for a connected medical device, and nearly every connected device now ships with a companion app.
The money is following the patients. Medicare spending on remote patient monitoring rose from $6.8 million in 2019 to $194.5 million in 2023, close to 30x in 4 years, per a 2025 Peterson Center on Healthcare analysis.
The installed base is already at platform scale. ResMed’s FY2025 annual filing reports more than 30 million patients on cloud-connected devices through AirView and over 10 million registered users on its myAir companion app.
Across every category, the companion app decides patient retention, clinical data quality, and reimbursement eligibility. That puts medical device companion app development on the product roadmap, with budget and headcount to match.
Four connected medical device categories account for most of the build activity we see: continuous glucose monitor (CGM), RPM, cardiac monitors, and smart inhalers. This guide takes each one in turn, covering FDA classification, Bluetooth Low Energy (BLE) architecture, HIPAA compliance, what the build costs, and how shipped products actually got cleared. Built for the team that has a wearable medical device and needs an app.
What does it take to build a companion app for a medical device?
A medical device companion app requires FDA classification assessment (non-device, accessory, or SaMD), BLE architecture validated against target hardware, HIPAA-compliant data infrastructure, and a QMS-aligned development process that produces both working software and the documentation package needed for 510(k) or De Novo submission. Requirements shift by device category: CGM, RPM, cardiac, and smart inhaler apps sit on different regulatory pathways and need different data architectures.
Key Takeaways:
- Your companion app carries its own FDA classification, and that classification sets your regulatory burden. Most teams assume the app inherits the hardware’s clearance. It doesn’t. An app that interprets, alerts, or recommends crosses from non-device into accessory or SaMD territory, and finding that out mid-build means rework.
- CGM, RPM, cardiac, and smart inhaler companion apps are four different products. They share BLE and HIPAA foundations, then diverge on FDA pathway, data architecture, liability profile, and clinical workflow integration. Each category gets its own section below, so skip to yours.
- The checklist and 5-phase process here are built for FDA submission. Shipping the software is one deliverable, and the submission package is the other. A working app that lacks QMS documentation, an SBOM, or a validated test suite can’t be submitted, so the process produces both together.
- Your app’s tier sets the budget. A connected-device app starts around $180,000 over 8 to 14 months before any regulatory work. HIPAA stacks one cost overlay on that base. For Tier 2 and Tier 3, the FDA layer (QMS, IEC 62304 documentation, verification and validation, submission fees) stacks a second. The cost section prices each tier.
Table of Contents:
- What is a medical device companion app, and how does FDA classify it?
- The medical device companion app matrix: FDA, HIPAA, BLE, and key challenges by device type
- Universal requirements: what every medical device companion app needs
- CGM companion app development (continuous glucose monitor)
- RPM companion app development (remote patient monitoring)
- Cardiac monitor companion app development (Holter, patch, ILR)
- Smart inhaler companion app development
- The development process for medical device companion apps
- How much does medical device companion app development cost?
- Real-world companion app examples: what got cleared, and why
- The complete companion app development checklist for medical device teams
- Why choose Topflight Apps for medical device companion app development
- What separates a compliant companion app from a costly rebuild
What is a medical device companion app, and how does FDA classify it?
A companion app for medical devices is a mobile application that connects to a hardware medical device, usually over Bluetooth Low Energy, to display, transmit, store, or analyze that device’s data, or to control its functions. FDA classifies the app on what the app does with the data. The hardware’s own clearance doesn’t carry over.
FDA’s September 2022 Policy for Device Software Functions and Mobile Medical Applications, issued through the FDA Digital Health Center of Excellence, created the three tiers relevant to companion apps. That document decides whether yours counts as an FDA medical device app at all. Which tier your app lands in shapes the submission path and the documentation load behind it, so settle it before you write code.

The same decision in text:
- The app only displays raw device data, exactly as received → Tier 1, non-device software. FDA doesn’t regulate the app itself.
- The app adds anything on top of display (alerts, trend interpretation, dosing recommendations, parameter changes) → Tier 2, accessory to the device. Plan for a 510(k) of its own or inclusion in the parent device’s submission.
- The app performs a medical function with no paired hardware at all → Tier 3, standalone SaMD, on its own regulatory pathway.
- Unsure which branch you’re on → ask FDA: a Q-Sub meeting for feedback, or a 513(g) request for a classification answer in writing.
Tier 1. Non-device software (no FDA oversight)
The app displays raw data from the device without interpretation: a CGM glucose reading on a phone screen exactly as the sensor sent it, with no trend analysis, alerts, or clinical decision support. FDA doesn’t regulate this tier as a medical device, and the app isn’t medical device software (MDSW). It’s a display.
Tier 1 exists on paper. The CGM example is empty in practice, because every commercial CGM app carries the sensor’s mandatory alarms, which puts shipped CGM apps inside the sensor’s clearance instead of Tier 1. More on that in the CGM section below.
Tier 2. Accessory to a medical device
The app supplements, augments, or controls the functions of a regulated medical device. FDA treats that as an accessory to a medical device and may require its own FDA 510(k) clearance, or inclusion in the parent device’s submission. An app that gives dosing recommendations from CGM data lands here. So does one that adjusts parameters on a cardiac rhythm monitor.
This is the tier that catches most companion app teams by surprise, and the most expensive one to fix once the build is underway.
The dosing example is also where FDA’s January 2026 Clinical Decision Support Software guidance lands. Non-device CDS has to be directed at a healthcare professional and can’t rest on analysis of signals or patterns. A patient-facing app recommending insulin doses from continuous glucose data fails both tests, so it’s device CDS under the new guidance exactly as it was under the 2022 version.
Tier 3. Standalone SaMD
The app performs medical device functions with no paired hardware at all, like using phone camera or microphone data to run a clinical assessment. That’s full Software as a Medical Device (SaMD), on the FDA SaMD track. SaMD classification follows your app’s own intended use, so when no predicate device exists to establish substantial equivalence, the route is the De Novo pathway and De Novo classification is what you come out with.
Related: SaMD App Development Guide
⚠ Regulatory Flag: The most common regulatory mistake in companion app development is building an app that qualifies as an accessory to a device (Tier 2), then submitting only the hardware for FDA clearance. That leaves the app’s risk classification unresolved, and when the app is integral to the device’s intended use, FDA expects both in the submission. Clarify it in an FDA pre-submission meeting (Q-Sub) before development starts. If you want the answer in writing, a 513(g) request for classification information has a published price: $7,820 at FY2026 rates, or $3,910 for a small business. Cheap next to finding out in month 9.
The medical device companion app matrix: FDA, HIPAA, BLE, and key challenges by device type
Here’s how the four connected medical device app categories compare across the dimensions that drive a build. If you’re at a medical device startup facing FDA for the first time, read the classification column first.
| Device Category | FDA Class | Companion App Classification | HIPAA Required? | Primary Protocol | Key Build Challenge |
|---|---|---|---|---|---|
| CGM (Dexcom G7, Abbott FreeStyle Libre) | Class II | Accessory, often needs its own 510(k) | Yes (PHI) | BLE GATT + proprietary SDK | Real-time glucose alerts; background sync reliability; insulin dosing liability if app gives recommendations |
| RPM Platform (blood pressure monitor, weight, pulse oximeter, SpO2) | Class II | Non-device display or accessory (depends on alerting logic) | Yes (PHI) | BLE + Wi-Fi; HL7 FHIR for EHR push | Multi-device aggregation; CPT codes 99453/99454 billing workflow; care team dashboard; alert fatigue management |
| Cardiac Monitor (Holter monitor, patch, implantable loop recorder) | Class II / III | Accessory, usually folded into the parent 510(k) or PMA | Yes (PHI) | BLE for patches; cellular for ILRs | Arrhythmia detection liability; ECG data fidelity; FDA cybersecurity requirements; clinician reporting workflow |
| Smart Inhaler (Propeller Health, Hailie/Adherium) | Class II | Accessory, often needs its own 510(k) | Yes (PHI) | BLE GATT | Medication event capture accuracy; geolocation correlation; care plan integration; pediatric UX requirements |
The four sections below take the categories one at a time, starting with CGM.

Universal requirements: what every medical device companion app needs
Four foundations apply across all four categories, ahead of anything device-specific. Get any of them wrong in any vertical and the result is the same: a failed FDA submission, a rejected App Store review, or a patient safety event.
BLE architecture and connection reliability
Medical device companion apps stand or fall on Bluetooth connection reliability. A dropped CGM reading or a missed cardiac event isn’t a UX problem. It’s a patient safety event. Four architecture decisions carry the most weight:
- BLE GATT profile selection: use standardized profiles (Heart Rate, Glucose) where available, and build custom GATT profiles only where the device requires proprietary data characteristics. Bluetooth pairing must be seamless across iOS and Android.
- Background data sync on iOS: the OS aggressively terminates background processes, so Core Bluetooth’s state preservation and restoration APIs have to be implemented correctly or readings are lost. This is where companion apps break most often.
- Reconnection logic: patients move, devices sleep, and the app has to reconnect gracefully without user intervention. Real-time data streaming needs robust retry logic and connection state management.
- Offline mode: readings get buffered locally and synced through a reliable data synchronization pipeline once connectivity resumes. No regulated IoT medical device can afford to lose data.
Battery impact belongs on the same list. Aggressive BLE polling drains the phone, and patients who charge more often use the app less, so battery optimization has to be designed into the connection architecture rather than patched in later.
For a deeper dive into BLE implementation patterns across iOS and Android, see our guide to BLE mobile app development.
HIPAA compliance
Every device in this matrix transmits protected health information (PHI), which makes medical device app HIPAA compliance mandatory for every companion app in these categories. Five requirements hold universally:
- Execute a business associate agreement (BAA) with your cloud provider, analytics vendor, push notifications service, and any EHR integration partner.
- Implement encryption at rest (AES-256) and encryption in transit (TLS 1.3) for all PHI.
- Audit logging: who accessed which patient record, when, and from which device. This is both a HIPAA requirement and an FDA expectation for regulated software.
- Minimum necessary data access: the software shouldn’t transmit more PHI than the clinical use case requires.
- Breach notification plan: the 60-day OCR notification window starts when a breach is discovered, not when it’s investigated.
Data privacy obligations reach past HIPAA once your app operates internationally or collects location data, and further still in pediatric populations. For a complete walkthrough, see our guide to HIPAA compliant app development.
FDA cybersecurity requirements (February 2026 guidance)
FDA cybersecurity guidance for medical devices applies to any device software, companion apps classified as accessories included. The current version is Cybersecurity in Medical Devices: Quality Management System Considerations and Content of Premarket Submissions, issued February 2026. It superseded the September 2023 edition most teams still cite, with a June 2025 version in between, and the retitle from “Quality System” to “Quality Management System” tracks FDA’s move to the QMSR. For a submission, four items are effectively mandatory:
- SBOM (software bill of materials): a complete inventory of all software components, including third-party libraries, open-source dependencies, and their known vulnerabilities.
- Vulnerability monitoring and patching plan: a documented process for responding to discovered vulnerabilities post-market.
- Penetration testing: required pre-submission for most device software.
- Secure development lifecycle (SDL) documentation.
⚠ Regulatory Flag: SBOM is the requirement most companion app teams are least prepared for, and the one with a statute behind it. For cyber devices, section 524B(b)(3) of the FD&C Act, added by FDORA in December 2022, makes the SBOM a legal requirement in premarket submissions, and the February 2026 guidance says so explicitly. Your React Native dependencies, your BLE parsing library, your analytics SDK: all of it has to be catalogued, versioned, and monitored for vulnerabilities. Build the SBOM process into development from day one instead of scrambling for it before submission.
HealthKit, Health Connect, and platform data sharing
Most medical device companion apps integrate with Apple HealthKit and Google Health Connect to share device data with the broader health ecosystem on iOS and Android. Both platforms run their own review in parallel with FDA:
- Apple requires HealthKit apps to have a clear clinical purpose, and wellness-only framing can trigger rejection.
- Health Connect requires data type permissions to be justified in the Play Store submission.
- Both platforms run enhanced review for apps handling sensitive health data categories (cardiac, glucose, respiratory).
Topflight Apps builds HIPAA-compliant, FDA-ready companion apps for medical device companies. Talk to our team about your device and intended use.
CGM companion app development (continuous glucose monitor)
| Quick Answer: On paper, a display-only CGM app is non-device. In practice the mandatory low-glucose alarm puts it inside the sensor’s iCGM clearance, and adding dosing logic turns it into a controller with its own special controls. |
Market Signal: Dexcom and Abbott dominate CGM hardware, but mid-market diabetes management app platforms and digital health platform companies are building companion layers on top of CGM data APIs. The Dexcom Real-Time API and the Abbott LibreLink developer program both open the door to third-party CGM companion app development, and that has grown into a real market for specialized development partners.
FDA classification for CGM companion apps
CGM companion apps occupy a narrow regulatory corridor. Display a glucose reading from a Dexcom G7 or Abbott FreeStyle Libre, and you’re generally non-device. Add trend-based insulin dosing recommendations, and you’re almost certainly an accessory that needs its own 510(k) or inclusion in the CGM system’s predicate submission. The dosing recommendation is the line, and the line is hard.
Key regulatory reference: the iCGM category comes from FDA’s March 2018 De Novo decision DEN170088, with special controls codified at 21 CFR 862.1355. Those controls govern interoperable CGM systems that share data with third-party apps. If your application ingests iCGM data, it may have to meet their interoperability requirements, which carry their own accuracy and connectivity validation standards.
Technical architecture: CGM
- Primary data source options: Dexcom Real-Time API (OAuth 2.0, REST), Abbott LibreLink Up API, or direct BLE from the sensor (needs an OEM partnership, and never attempt hardware reverse engineering).
- Real-time versus retrospective: the split that shapes CGM app development. Direct BLE to the transmitter is the only path to live readings and alerts. The public cloud APIs are retrospective by design and unfit for alerting. Teams get this decision wrong more than any other.
- Glucose alert logic: real-time alerts for high and low readings are the core patient safety feature. Build them with background processing, sound alerts, and Apple Watch / Wear OS mirroring, which is where wearable app development expertise starts to matter.
- Trend arrow display: FDA treats the trend arrow as clinically significant, so your UI has to match the cleared device’s trend arrow definitions exactly. Deviations create regulatory risk.
- Data latency: the Dexcom API runs up to a 3-minute delay. Your architecture has to handle that gracefully and never imply real-time accuracy it doesn’t have, so the cloud data pipeline accounts for the lag in every user-facing display.
- Insulin-on-board and carb tracking: add these features and you’ve crossed into clinical decision support territory, where FDA classification review comes before development.
CGM-specific HIPAA considerations
CGM data runs unusually deep. It reveals daily patterns of life, eating behavior, and activity level, and it can imply conditions beyond diabetes. The minimum necessary principle matters more here than almost anywhere: don’t transmit raw CGM time-series data to analytics platforms unless the clinical use case requires it.
⚠ Regulatory Flag: Several CGM companion app companies have drawn FTC warning letters for selling anonymized glucose data to third parties. Anonymizing CGM time-series data is technically difficult and legally contested. Default to this: collect what you need clinically, retain it for the minimum required period, never sell. The same holds for any mHealth app handling continuous physiologic data.
For teams building AI-driven analytics on top of CGM data pipelines, how AI is reshaping EHR workflows is useful context. See How will AI help change EHR?
RPM companion app development (remote patient monitoring)
| Quick Answer: An RPM app that aggregates and forwards readings is generally non-device, and interpretation or clinical alerting escalates it. The tighter constraint is usually CMS: CPT codes 99453/99454 documentation gates reimbursement whether or not FDA is involved. |
Market Signal: RPM reimbursement via CMS CPT codes 99453, 99454, 99457, and 99458 has built an enterprise B2B market for RPM platforms, and health systems are procuring RPM solutions in volume. The RPM companion app, the patient-facing component, is the highest-friction element in RPM adoption. Patients who don’t open the app generate no billable data. No data, no reimbursement, no program.
FDA classification for RPM companion apps
RPM companion apps span a wide classification range. A pure data display app showing blood pressure readings from a connected cuff is generally non-device. Software that generates alerts, scores clinical risk, or gives care recommendations from aggregated data is an accessory or SaMD. Most enterprise RPM platforms carry some alerting logic, which puts them into regulatory assessment right away.
Related: RPM app development
Technical architecture: RPM
- Multi-device management: enterprise RPM apps aggregate data from blood pressure monitors, pulse oximeters, weight scales, glucometers, and thermometers, often across manufacturers. Your BLE layer has to handle heterogeneous device profiles, which puts this much closer to IoT app development than to a single-device companion app.
- FHIR API integration: RPM data has to flow to the ordering provider’s EHR over HL7 FHIR R4. Epic, Cerner, and Athenahealth all have RPM integration pathways. Our Epic data integration guide details the USCDI on FHIR APIs and per-site approvals that pathway involves. This is the technical layer that makes EHR integration and health system partnership deals possible. For teams weighing EHR build decisions, see our guide on how to build an EHR system.
- Care team dashboard: the companion app is the patient side. You also need a clinician-facing dashboard for alert review, trend analysis, and documentation inside the clinical workflow. That’s two products, not one.
- CPT billing workflow: RPM reimbursement runs on CPT codes 99453/99454 for device setup and data supply, which means documented physiologic data collection for 16+ days per month plus documented clinical review time. Your platform generates the audit trail for billing. That audit trail isn’t a feature, it’s the business model.
- Alert fatigue architecture: RPM generates enormous data volume, so your alerting logic has to be tunable by clinical protocol rather than a binary on/off. Care teams that can’t tune it will switch alerts off entirely, and that takes patient engagement and the program’s clinical value down with it.
RPM-specific compliance considerations
RPM puts a covered entity, the ordering health system, in the seat of your direct customer. That makes HIPAA compliance non-negotiable and the BAA a sales prerequisite rather than an afterthought. Health system procurement teams will want a completed HIPAA security assessment and BAA before any pilot. The billing side carries just as much weight: see our overview of EHR in medical billing for how RPM data flows into reimbursement.
Building an RPM platform and need FHIR-based EHR integration alongside the patient app? Topflight Apps handles both layers.
Cardiac monitor companion app development (Holter, patch, ILR)
| Quick Answer: The regulated function in cardiac monitoring is the arrhythmia interpretation, not the app. Keep the ECG out of the patient app and the app stays non-device. Interpret on the phone and you’re building SaMD. |
Market Signal: Cardiac monitoring is the highest-stakes companion app category. Class III implantable devices like implantable loop recorders come with companion apps that have to meet the same regulatory rigor as the device itself.
The cardiac monitor companion app market sits inside the arrhythmia monitoring devices industry, valued at $9.1 billion as of 2026 per Global Market Insights, and pushed higher by demand for atrial fibrillation detection and the patient awareness of heart rhythm irregularities that Apple Watch has driven.
FDA classification for cardiac companion apps
Cardiac companion apps are almost always classified as accessories to a medical device or folded into the parent device’s FDA submission. A display-only ECG viewer is potentially non-device. Once the software detects, classifies, or alerts on arrhythmias, it’s an accessory or SaMD. For Class III devices (implantable loop recorders), the companion app usually rides in the PMA submission, a different regulatory pathway from 510(k).
Key regulatory note: patient-facing arrhythmia detection is governed by two classification regulations, 21 CFR 870.2345 (electrocardiograph software for over-the-counter use, product code QDA) and 21 CFR 870.2790 (photoplethysmograph analysis software for over-the-counter use, QDB), both created through FDA’s September 2018 De Novo orders. Each states the device is not intended to provide a diagnosis: the cleared claim is notifying the user of a possible irregular rhythm, and the diagnosis stays with a clinician. Their special controls require clinical performance testing of the detection algorithm, so any app running algorithm-based arrhythmia detection needs validated clinical data behind it.
Technical architecture: cardiac
- ECG fidelity requirements: cardiac companion apps have to transmit ECG waveforms without signal degradation, and any compression algorithm gets validated against clinical accuracy standards. This isn’t a general-purpose data compression problem.
- Transmission protocols: the phone is optional in some architectures. iRhythm’s connected Zio moves data from patch to a dedicated gateway over Bluetooth, then from gateway to cloud over cellular, so the patient’s phone sits outside the ECG path and handles only symptom logging. Other ambulatory Holter monitor patches use BLE to the phone, then cellular or Wi-Fi upload. Either way, the full data pathway has to be validated for signal integrity from sensor to clinician report.
- ILR remote programming: apps for implantable loop recorders may include remote parameter adjustment, which carries more regulatory risk than any other companion app function and needs deep OEM partnership. See our guide on heart monitoring app development for additional technical context.
- Clinician reporting workflow: cardiac companion apps typically generate PDF reports for the ordering cardiologist. Report format, terminology, and clinical completeness fall under the same FDA submission that covers the device.
- Real-time alerting: atrial fibrillation and critical arrhythmia alerts have to reach patients and care teams at a validated latency. The alerting architecture is part of the device’s intended use claim, so it gets tested accordingly.
⚠ Regulatory Flag: Cardiac companion apps carry the heaviest legal exposure of any device category. A missed arrhythmia, whether from a BLE disconnect, a background sync failure, or a software bug, becomes a potential wrongful-death event rather than a mere product defect. For cardiac, Quality Management System (QMS) documentation, validated testing protocols, and post-market surveillance aren’t line items you can defer. They’re existential.
Cardiac-specific cybersecurity requirements
FDA’s February 2026 cybersecurity guidance lands with particular force on cardiac device software: remote access vulnerabilities in cardiac devices have already produced voluntary recalls, which is exactly the class of failure the guidance exists to prevent. Your app’s authentication, session management, and network communication have to satisfy the cybersecurity section of the 510(k) or PMA submission. AI in healthcare compliance matters more here every year, as manufacturers explore automated vulnerability detection in post-market surveillance.
Smart inhaler companion app development
| Quick Answer: Logging inhaler actuations is a data function. What turns a smart inhaler system into a device is the claim set, adherence improvement or exacerbation prediction, which pulls sensor and app into a Class II clearance together. |
Market Signal: Asthma and COPD together affect well over 500 million people worldwide, per WHO figures (asthma alone: 363 million as of 2023). Propeller Health (acquired by ResMed), Hailie (Adherium), and Teva’s Digihaler are the established players.
Third-party smart inhaler app development is growing as payers and pharmaceutical companies go after medication adherence data to justify reimbursement and demonstrate real-world drug efficacy.
FDA classification for smart inhaler apps
Smart inhaler companion apps typically qualify as accessories to the inhalation device. FDA has issued specific guidance on combination products, meaning devices that pair drug delivery with digital components, and it applies directly to smart inhalers. The companion app may need its own 510(k) clearance, or it may ride the combination product submission, depending on the OEM’s regulatory strategy.
Technical architecture: smart inhaler
- Medication event capture: the core function is detecting an inhaler actuation and logging the time, location, and device ID. BLE GATT profiles for inhaler event capture aren’t standardized, so most need device-specific SDK integration, unlike the standardized profiles you get for heart rate or glucose.
- Geolocation correlation: smart inhaler apps correlate medication events with location (home, school, outdoors) and environmental data (pollen count, air quality index). That combination raises privacy considerations you don’t see elsewhere, especially for pediatric users.
- Adherence scoring: apps that score adherence and present the results to care teams or payers cross from display into clinical decision support, and FDA classification review comes before the feature ships.
- Pediatric UX requirements: a large share of smart inhaler users are children, so the software has to serve both the child (simple, visual, gamified) and the parent (detailed adherence data, care plan access). Two UX modes in one app.
- Care plan integration: smart inhaler apps increasingly tie into asthma action plans. When the app hands out escalation guidance (“take rescue inhaler now,” “call your doctor”), that escalation logic needs regulatory review, because it counts as clinical decision support.
Smart inhaler: pharma partnership considerations
Several smart inhaler companion app projects are funded by pharmaceutical companies after real-world adherence data for their drugs. That sets up a distinct data governance requirement: patient inhaler usage data may be valuable to the drug manufacturer, but sharing it takes explicit, granular informed consent, and it invites FTC scrutiny if the data sharing isn’t clearly disclosed.
The broader cost of AI in healthcare matters here, since pharma partners now tend to expect AI-driven adherence analytics as part of the companion app deliverable.
Other device categories: insulin pumps, hearing aids, neurostimulation, and CPAP
One rule covers the device categories this guide doesn’t break out in full: the app’s tier turns on whether it displays data or commands therapy.
| Device Category | Typical App Tier | What Escalates It | Cleared Example | Closest Archetype |
|---|---|---|---|---|
| Insulin pump | Tier 2 accessory; Tier 3 if the app owns the dosing algorithm | Phone-initiated bolus | Tandem t:connect, K203234 (Feb 2022) | CGM |
| Hearing aid | Tier 2 accessory; Tier 3 when the software is the hearing aid | Self-fitting: deriving amplification from a hearing test | Apple Hearing Aid Feature, DEN230081 (Sept 2024) | Smart inhaler |
| Neurostimulation | Tier 2 accessory riding the parent PMA as a supplement | Remote programming of stimulation parameters | Abbott patient controller app with NeuroSphere Virtual Clinic (2021) | Cardiac monitor |
| CPAP / PAP | Tier 1 non-device for adherence data | Adding any clinical or diagnostic function | ResMed myAir, K241216 (2024) | RPM |
Insulin pumps
FDA split automated insulin delivery into interoperable pieces, and the app’s tier follows what it does with them. Displaying pump status is unregulated. Programming a bolus from the phone needs its own clearance. Owning the dosing algorithm makes the app a controller under its own special controls. Nearest to CGM, except this app can overdose a patient.
Hearing aids
The 2022 OTC final rule reset the category. Volume control is accessory work, but self-fitting, where the user derives their own amplification curve from a hearing test, makes the software the device. Apple’s 2024 authorization was the first software-only hearing aid. Closest to the smart inhaler model, though the therapy runs continuously and in real time.
Neurostimulation
Implantable stimulators are mostly Class III, so the patient controller app rides the parent PMA as a supplement instead of filing its own 510(k). Remote programming is where the engineering gets hard: authenticating and bounding therapy changes pushed to an implant over cloud and BLE. It maps to cardiac monitors, with one difference: this app writes settings back to the implant rather than only reading from it.
CPAP (continuous positive airway pressure)
Adherence tracking sits below FDA’s device threshold, so the binding constraint is CMS rather than FDA. Medicare’s PAP policy defines adherence as four hours a night on 70% of nights across a 30-day stretch in the first three months, which makes the compliance report the actual product. Nearest to RPM, and for the same reason: payer reporting drives the build. The scale point lands here too: myAir’s 10+ million registered users (ResMed FY2025 filing) make it the largest companion-app installed base in this guide, and its adherence tracking still sits below FDA’s device line. Scale and regulatory burden are unrelated.
The development process for medical device companion apps
An FDA companion app medical device teams can submit with confidence is a different exercise from a consumer health app. The process has to turn out two things at once: the software clinical teams want and the documentation FDA requires. Here’s the timeline and phase structure Topflight Apps uses across all four device categories.
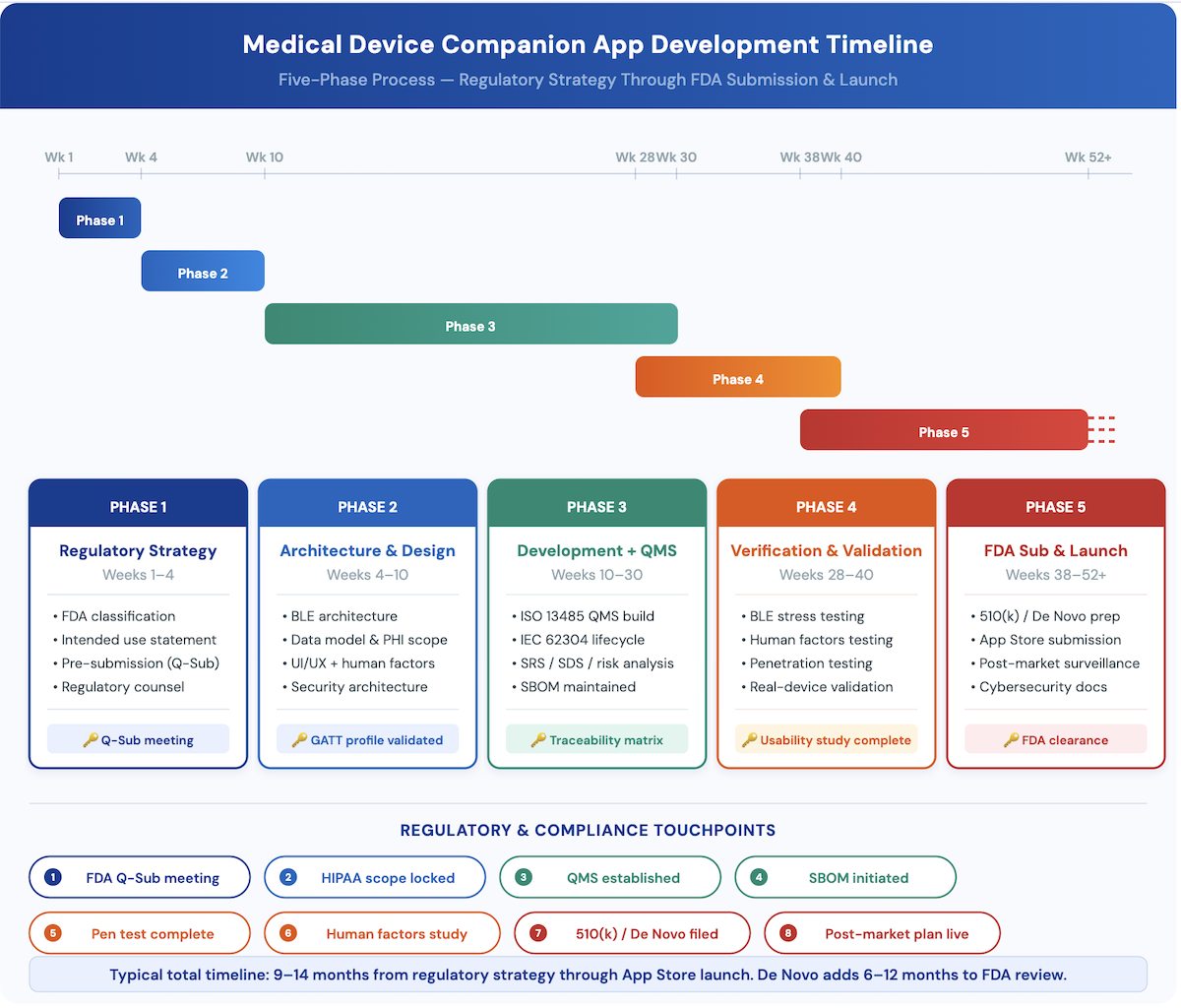
| Phase | Weeks | Focus |
|---|---|---|
| Phase 1 | Weeks 1–4 | Regulatory strategy: classification, intended use, Q-Sub |
| Phase 2 | Weeks 4–10 | Architecture and design: BLE, data model, UI/UX, security |
| Phase 3 | Weeks 10–30 | Development with QMS documentation |
| Phase 4 | Weeks 28–40 | Verification and validation |
| Phase 5 | Weeks 38–52+ | FDA submission and app store launch |
Phase 1: Regulatory strategy (weeks 1–4)
- Determine companion app FDA classification from intended use, and bring in regulatory counsel early.
- If the app is an accessory or SaMD, schedule an FDA pre-submission (Q-Sub) meeting to align on pathway before you sink real money into development.
- Document the intended use statement and indications for use. These drive every design decision that follows.
- Decide whether the app goes into the parent device submission or files separately.
Phase 2: Architecture and design (weeks 4–10)
- BLE architecture design: device pairing, GATT profile selection, connection management, and background sync for both iOS and Android.
- Data model design: what PHI you collect, where it lives, retention periods, access controls.
- UI/UX design for the patient-facing and clinician-facing interfaces, plus accessibility (Section 508 compliance for any federally funded program). Human factors engineering documentation starts here.
- Security architecture: authentication, encryption, session management, and the SBOM foundation.
Phase 3: Development with QMS documentation (weeks 10–30)
- Development runs under an ISO 13485:2016 quality management system. Since February 2, 2026 that alignment is FDA’s own rule: the Quality Management System Regulation (QMSR) incorporates ISO 13485:2016 by reference. Software development lifecycle documentation is required for every FDA submission.
- IEC 62304 software lifecycle compliance: risk classification of software units, and a traceability matrix running from requirements to design to test cases.
- Concurrent documentation: the software requirements specification (SRS), software design specification (SDS), and risk analysis per ISO 14971.
- SBOM generation, maintained throughout development rather than bolted on after the build.
- Compliance with 21 CFR Part 820, now the Quality Management System Regulation (QMSR), where it applies.
Phase 4: Verification and validation (weeks 28–40)
- Verification testing: unit tests, integration tests, BLE connection stress testing, data integrity validation.
- Validation testing with representative users. FDA requires human factors studies for many device categories.
- Penetration testing, required for the FDA cybersecurity submission documentation.
- Performance testing on real target iOS and Android hardware with the actual BLE peripherals. Emulators don’t cut it for regulated software.
Phase 5: FDA submission and app store launch (weeks 38–52+)
- 510(k) or De Novo preparation: the software documentation package, including SRS, SDS, hazard analysis, V&V testing summary, SBOM, and cybersecurity documentation.
- Apple App Store and Google Play submission. Medical device apps get enhanced review, so plan for a 2–4 week review timeline.
- Post-market surveillance plan: a documented process for monitoring adverse events, software anomalies, and cybersecurity vulnerabilities after launch.
How much does medical device companion app development cost?
Medical device companion app development cost is set by your app’s FDA tier before it’s set by your feature list. The base is published: a hardware-connected app starts around $180,000 and takes 8 to 14 months, with the time going to BLE pairing, firmware-and-app coordination, OTA update flows, and certification cycles (figures from our app development cost guide). HIPAA stacks one overlay on that base, and the FDA layer stacks a second. Here’s the same app priced at all three tiers.
Cost by FDA classification tier
| Tier | Regulatory Path | Cost Structure |
|---|---|---|
| Tier 1: non-device display | No FDA submission | Base build from about $180,000 over 8 to 14 months. Add the HIPAA overlay if PHI is in scope: roughly $5,000 to $25,000 at pilot stage, $40,000 to $100,000 for compliance credible to enterprise buyers. |
| Tier 2: accessory with 510(k) | 510(k) clearance | Everything in Tier 1, plus the FDA layer: QMS work under the QMSR, IEC 62304 documentation, verification and validation, and the $26,067 510(k) fee ($6,517 small business) at FY2026 rates. |
| Tier 3: standalone SaMD | De Novo or PMA | Everything in Tier 2 at higher intensity, plus clinical evidence and post-market obligations. De Novo runs $173,782 ($43,446 small business) at FY2026 rates. |
FDA’s user fees are exact and public, and almost no cost article prints them. FY2026 rates below, from Federal Register notice 90 FR 35895 (July 2025). FDA resets these every October 1, and FY2027 rates land in an August notice, so check the year before you budget off this table.
| Submission | Standard Fee (FY2026) | Small Business Fee (FY2026) |
|---|---|---|
| 510(k) premarket notification | $26,067 | $6,517 |
| De Novo classification request | $173,782 | $43,446 |
| 513(g) request for classification information | $7,820 | $3,910 |
| Premarket approval (PMA) | $579,272 | $144,818 |
| Annual establishment registration | $11,423 | $11,423 (no reduction) |
Three mechanics decide what you actually pay. Small business status covers companies with $100 million or less in gross receipts, which is nearly everyone reading this, and cuts most submission fees by 75%. File the Small Business Request at least 60 days before your submission, because if you miss the window the same 510(k) costs 4 times more, and the status expires every September 30, so requalify annually. And a 510(k) routed through an FDA-accredited third-party reviewer carries no user fee at all.
Two more numbers put the table in proportion. FDA collects fees on roughly 3,850 510(k)s a year against about 81 De Novo requests, so if someone tells you your companion app needs a De Novo, you’re being placed in the 2% case and should hear the reasoning. And the cheapest line in the table may be the most valuable: a 513(g) request buys FDA’s written answer on your classification for $7,820 ($3,910 small business). Teams routinely discover their tier too late, and there’s a published price for finding out early.
What increases companion app budgets
The FDA layer has no honest single price, so treat any page that quotes one with suspicion. Three components drive it, and none has a rate card. Quality system setup and registrar audits come first: registrars don’t publish rates, and since the QMSR took effect in February 2026 this line stopped being optional for regulated apps. IEC 62304 documentation is next, and its burden scales with the software safety class, so a Class A display app and a Class C dosing app carry very different loads from the same codebase. Verification, validation, and human factors work round it out, driven by participant counts and test protocols rather than a flat rate.
Recurring costs are the ones budgets miss. Establishment registration is $11,423 every year with no small business rate (FY2026 added a discretionary hardship waiver for firms at $1 million or less in gross receipts). Maintenance runs about 25% of the build per year, and for a regulated product that line covers compliance refresh, SBOM upkeep (statutory for cyber devices), and pen-test cycles, not just bug fixes. At the platform end, our cost guide stages the security and compliance overlay from $5,000 at MVP up past $400,000 for a regulated multi-customer platform.
One more point, because it cuts against what other pages promise: AI-era savings land on routine implementation, while regulated, safety-critical work still commands senior rates. Documentation, validation, and regulatory review don’t compress. So the AI discount applies to a smaller share of a companion app budget than a consumer app budget, and a device founder pricing off AI-savings claims built for consumer apps is being quietly misled.
Real-world companion app examples: what got cleared, and why
Bluetooth medical device app development looks abstract until you read actual clearances. The four products below all pair a BLE device with a shipped companion app, and the hardware’s class predicts nothing about where each app landed: the spectrum runs from software FDA never touched to software that is the device. The clearance numbers are just the proof.
| Product | App Tier | Deciding Capability | Reference |
|---|---|---|---|
| Dexcom G7 app | Tier 2, cleared inside the iCGM submission | Primary display with a non-defeatable low-glucose alarm | QBJ, 21 CFR 862.1355; K213919 |
| Dexcom Follow | Tier 2, lower risk | Labeled not for treatment decisions, passive viewing only | Within Dexcom’s CGM clearances |
| iRhythm MyZio | Tier 1 | Symptom diary. Never touches the ECG | No clearance for the app itself |
| Propeller | Tier 2 accessory system | Adherence and exacerbation-prediction claims | CAF, 21 CFR 868.5630; K161454 |
| Omnipod 5 app | Tier 3 controller | Calculates and commands insulin dosing | QJI, 21 CFR 862.1356; K203774 |
Dexcom G7
The G7 app is part of the regulated system, not a viewer sitting outside it. It’s a primary display, and its urgent low alarm fires at 55 mg/dL and can’t be turned off or adjusted. A mandatory clinical alarm is a device function, which is why the app was cleared inside the iCGM submission rather than separately. Then look at Dexcom Follow, the app built for parents and partners: its labeling states that treatment decisions shouldn’t be made from it, and that one sentence is what keeps it a lower-risk secondary display. Same data, same company, different regulatory weight, decided by what the app is permitted to be used for. The scale behind that labeling sentence: $4.03 billion in FY2024 revenue and 2.8 million customers, per Dexcom’s investor materials.
iRhythm MyZio
The most instructive app in cardiac monitoring is the one that barely does anything. MyZio is a symptom diary. It doesn’t capture, display, or transmit ECG. On the connected Zio AT, data moves from the patch to a dedicated gateway to iRhythm’s cloud, and the patient’s phone isn’t in that path at all. The arrhythmia intelligence lives in ZEUS, iRhythm’s deep-learning analysis software, cleared as standalone SaMD in February 2023, with output reviewed by certified technicians before a clinician sees it.
iRhythm put the regulated function where it could be controlled and left the phone outside the device boundary. That’s an architecture decision, not an accident. The company’s FY2024 filing shows the stakes: 6 million patients served and 1.8 billion hours of curated heartbeat data, a volume of ECG analysis that belongs in separately cleared software, not on a phone.
Propeller Health
Propeller’s sensor and app are cleared together as a Class II system, under a nebulizer product code, with more than eight clearances since 2012 covering individual inhalers from GSK, Boehringer Ingelheim, and AstraZeneca. Logging actuations on its own would be a data function. What makes this a device is the claim set: Propeller records puffs and then goes further, making clinical claims about adherence and exacerbation prediction.
The claims are what convert a logger into a regulated product. One absence worth noting: Propeller reports no standalone user count anywhere, since it sits inside ResMed’s segment reporting, which is why this teardown carries no scale figure.
Omnipod 5
Insulet’s app sits at the far end: it hosts the algorithm that decides insulin delivery. FDA broke the system into interoperable blocks, and the controller carries its own special controls as an interoperable automated glycemic controller. One detail worth copying: the algorithm runs on the Pod, not the phone, so automated delivery continues when the phone is out of range.
A trend arrow or a follower notification doesn’t cross the controller line. A dosing command does. Adoption there is real: roughly 500,000 active Omnipod customers as of FY2024, about 365,000 of them on Omnipod 5.
The complete companion app development checklist for medical device teams
This checklist maps to the five-phase process above. Use it as a readiness assessment before you bring in a medical device app development partner or kick off an internal build.

Regulatory readiness
- ☐ Companion app intended use statement documented
- ☐ FDA classification determined: non-device / accessory / standalone SaMD
- ☐ If accessory or SaMD: FDA Q-Sub (pre-submission) meeting completed
- ☐ Device combination product strategy determined if applicable
- ☐ Regulatory counsel engaged
Quality Management System
- ☐ ISO 13485:2016 QMS in place or gap analysis completed (the QMSR baseline since February 2026)
- ☐ IEC 62304 software lifecycle documentation structure established
- ☐ Software Requirements Specification (SRS) drafted
- ☐ Risk management file started (ISO 14971)
- ☐ Traceability matrix: requirements → design → test cases
Technical architecture
- ☐ BLE GATT profile selected and validated against target hardware
- ☐ Background sync architecture designed for both iOS and Android
- ☐ Offline data buffering implemented
- ☐ HealthKit and Health Connect integration scoped
- ☐ FHIR R4 integration designed if EHR connection required
- ☐ Multi-device management architecture designed if RPM use case
HIPAA and data security
- ☐ HIPAA applicability determination documented
- ☐ BAAs executed: cloud provider, analytics, push notifications, EHR integration
- ☐ Encryption at rest and in transit confirmed for all PHI
- ☐ Audit logging implemented
- ☐ Data retention policy documented
- ☐ SBOM generation process established
FDA cybersecurity
- ☐ SBOM complete and versioned
- ☐ Penetration testing completed
- ☐ Vulnerability disclosure policy documented
- ☐ Post-market patching plan documented
- ☐ Authentication and session management reviewed against OWASP Mobile Top 10
Verification, validation, and launch
- ☐ BLE connection stress testing completed on physical target devices
- ☐ Human factors usability testing completed with representative users
- ☐ 510(k) or De Novo software documentation package prepared
- ☐ Apple App Store and Google Play medical device category submission prepared
- ☐ Post-market surveillance plan documented and assigned
Need a development partner who understands both the build and the regulatory documentation? Topflight Apps has shipped FDA-ready companion apps for device companies across all four categories above.
Why choose Topflight Apps for medical device companion app development
Topflight Apps builds companion apps for medical device companies: FDA-classified accessories, RPM platforms, and connected device interfaces. Picking a medical device software development company comes down to two proofs: that the firm understands the reimbursement and regulatory plumbing, and that it has shipped regulated, device-connected software. Here are ours.
- FDA-ready development: QMS-aligned process, IEC 62304 documentation, and SBOM generation built into the development lifecycle.
- BLE expertise across CGM, RPM, cardiac, and respiratory device categories, tested on physical hardware rather than emulators.
- HIPAA-compliant architecture from day one: BAA management, encryption, audit logging.
- FHIR-based EHR integration for RPM billing workflows and health system partnerships.
- Apple HealthKit and Google Health Connect integration, including enhanced App Store review preparation.
- Human factors and usability testing for FDA submissions.
Proof from the portfolio
Dedica Health: the reimbursement proof. Dedica’s remote patient monitoring platform monitors 1,100+ patients daily, runs as SaaS at $300,000 in annual recurring revenue, and keeps more than 80% of patients hitting their CPT code targets. That last number is the one an RPM program can’t survive without, and it’s the same constraint the RPM section above calls the binding one. The scoping decision is as instructive as the outcome: the first release deliberately skipped a consumer mobile app, taking patient reports over SMS and sensor data through an aggregation API, because that was the shortest path to billable, documented data. Reimbursement fluency is a build skill, and it transfers directly to companion app work.
Joovv: the regulated companion app proof. Joovv’s BLE companion app for connected light therapy hardware is the artifact this guide describes, built by us: documented to IEC 62304, HIPAA compliant, pulling from HealthKit and Google Fit, with background processing and session recovery designed in. It also carries the unglamorous requirement that separates device work from consumer work: supporting legacy hardware alongside current models, which meant recovering BLE commands from the original codebase and reimplementing them cleanly. Light therapy sits outside the four categories above; the engineering does not.
From the delivery record
Three field notes from our engineering leads:
Our BLE practice runs one design rule that doubles as regulatory advice: expose a single control characteristic for commands and a separate notify characteristic for telemetry. The capability that escalates an app’s FDA tier is commanding therapy rather than displaying data, and a control characteristic that writes therapy parameters is exactly that capability, expressed in the protocol. The GATT contract your team designs in week 3 determines the FDA tier you discover in month 9.
Our delivery leads plan firmware and app development as one collaborative track. When the firmware is still moving under the app, debugging has to be joint and often physically co-located, so sprints get scheduled that way from the start.
And our architects settle PHI scope before architecture. On one connected-device build the hardware collected no PHI, so a non-HIPAA stack was the deliberate choice: a regulatory decision made before any technical one, which is the sequencing this whole guide argues for.
Tell us about your device and intended use. We’ll scope your companion app and flag the regulatory considerations specific to your category.
What separates a compliant companion app from a costly rebuild
A medical device companion app is a regulated software product, not a generic mobile app with Bluetooth bolted on. It has to be built with the same rigor as the hardware it accompanies: documented, validated, and defensible to FDA, patients, and clinicians.
The four categories here, CGM, RPM, cardiac, and smart inhaler, each carry their own regulatory classification risks, BLE architecture requirements, and compliance obligations. Sorting those distinctions out before development begins is the difference between a clean FDA submission and a major redesign mid-clearance.
The development process and checklist above are your starting point. Settle your regulatory strategy before your architecture, and stand up your compliance infrastructure before the first patient data flows through the system. Every companion app for medical device hardware starts with the same question: what does FDA expect from this software? The teams that answer it early are the ones that ship.
Frequently Asked Questions
Does a companion app for a medical device need FDA clearance?
It depends on what the app does. An app that only displays raw device data typically doesn’t need its own clearance. An app that interprets data, gives clinical recommendations, or controls the device is classified as an accessory or SaMD, and it likely needs 510(k) clearance, De Novo classification, or inclusion in the parent device’s submission.
What is the difference between a medical device companion app and a standalone health app?
A companion app is tethered to a specific hardware device and exists to extend, display, or transmit that device’s data. A standalone health app runs on its own using phone sensors or manual input. FDA regulates companion apps by the device relationship; standalone health and wellness apps can fall outside FDA oversight entirely unless they perform SaMD functions.
How long does it take to build and get FDA clearance for a companion app?
Typical timeline: 9–14 months from regulatory strategy through App Store launch, with wide variation by device category and FDA pathway. Phase 1 (regulatory strategy) takes 4 weeks minimum, the 510(k) review itself adds 3–6 months, and De Novo adds 6–12 months. The most common delay is discovering regulatory requirements mid-development, which forces architecture rework.
What is IEC 62304 and does it apply to my companion app?
IEC 62304 is the international standard for medical device software lifecycle processes. It applies to any software that’s part of a medical device or is itself classified as one, including companion apps classified as accessories. It requires documented software development planning, risk classification of software units, and traceability from requirements through verification.
Does a CGM companion app need its own 510(k)?
If the app provides trend analysis, dosing guidance, or clinical alerts beyond raw data display, FDA is likely to classify it as an accessory that needs its own 510(k) or inclusion in the CGM system’s submission. The iCGM special controls at 21 CFR 862.1355 impose extra requirements on apps that connect to interoperable CGM systems. Clarify classification with FDA in a pre-submission meeting before development.
What BLE protocols do medical device companion apps use?
Most use Bluetooth Low Energy (BLE) with GATT profiles: standardized where available (Glucose, Heart Rate, Blood Pressure) and custom where device-specific data characteristics are needed. BLE GATT is the dominant protocol for short-range device communication. Some cardiac ILR systems use cellular connectivity for continuous cloud upload instead of BLE or alongside it.
What does an SBOM need to include for a medical device companion app?
For cyber devices, an SBOM is a statutory requirement under section 524B(b)(3) of the FD&C Act, added by FDORA and effective March 2023, and FDA’s February 2026 cybersecurity guidance makes that explicit. The SBOM has to list all commercial, open-source, and off-the-shelf software components, their version numbers, and known vulnerabilities. That covers platform frameworks, BLE libraries, analytics SDKs, cloud service dependencies, and any third-party modules, and it has to be maintained and versioned throughout the product lifecycle.
Can I use a third-party CGM API (Dexcom, Abbott) in my companion app?
Yes. Dexcom offers a Real-Time API (OAuth 2.0, REST) and Abbott offers the LibreLink Up API, and both enable third-party companion app development. Two constraints shape what you can build. First, the public API data is delayed by design: the delay was set with regional regulators specifically to keep real-time clinical decisions from being made off API data. Real-time access runs through Dexcom’s Partner Web APIs, separately cleared in July 2021 and invite-only, and live glucose readings with alerts mean talking to the transmitter directly over BLE. Second, using these APIs doesn’t exempt your app from FDA classification review: if your app adds clinical logic on top of the CGM data, the app itself may need regulatory clearance no matter where the data comes from.
What is the difference between RPM companion app requirements and general mHealth app requirements?
RPM companion apps have to support CMS billing compliance (CPT 99453/99454 documentation), multi-device aggregation, EHR integration via FHIR, and care team alerting workflows. General mHealth apps typically lack these enterprise clinical workflow requirements. RPM apps also need BAAs with health system customers as a procurement prerequisite, not just a compliance checkbox.
How do I handle HIPAA compliance for a companion app that works with an implantable cardiac device?
The same HIPAA requirements apply (encryption, BAAs, audit logging, breach notification), but cardiac devices add heightened cybersecurity requirements, because unauthorized access carries direct patient safety implications. FDA’s cybersecurity guidance explicitly references cardiac device vulnerabilities. Integrate your HIPAA compliance program with your FDA cybersecurity submission rather than running it as a separate workstream.
How much does it cost to develop a medical device companion app?
A companion app for a connected medical device starts around $180,000 and takes 8 to 14 months, and that’s before any regulatory work. The time goes to BLE pairing, firmware and app coordination, OTA update flows, and certification cycles. What moves the number is your app’s tier, not your hardware’s. A Tier 1 display app pays the base cost plus HIPAA work if PHI is in scope, which runs from about $5,000 at pilot stage to $40,000 to $100,000 for a version credible to enterprise buyers. A Tier 2 accessory adds the FDA layer on top: quality system work, now that ISO 13485 conformity is the Part 820 requirement rather than a nice-to-have, plus IEC 62304 documentation, verification and validation, and a 510(k) fee of $26,067, or $6,517 if you qualify as a small business, at FY2026 rates. Tier 3 SaMD adds clinical evidence and post-market obligations on top of that. Two costs teams routinely miss: the annual establishment registration fee of $11,423, which recurs every year and has no small business rate, and maintenance at about 25% of build per year, which for a regulated product means compliance refresh, SBOM upkeep, and pen-test cycles, not just bug fixes. Full breakdown in the cost section above and in our <a href=”https://topflightapps.com/ideas/app-development-costs/”>app development cost guide</a>.
Do I need IEC 62304 compliance if my app only displays device data?
No, and the more useful correction is that you never legally need it in the US, even for a regulated app. IEC 62304 is an FDA-recognized consensus standard, not a regulation. Plenty of agency pages claim FDA requires IEC 62304, and that’s false. Recognition means you may declare conformity to it in a 510(k), De Novo, or PMA, which streamlines review by letting reviewers rely on the standard’s process framework instead of assessing your lifecycle from scratch. What’s actually mandatory is FDA’s software documentation expectations for whatever submission you’re making, and a genuine Tier 1 app makes no submission at all. So the answer is no in the legal sense and yes in the practical one, for three reasons. The display-only boundary is thinner than it looks, and a single alert crosses it. Device partners and health systems ask about lifecycle documentation during supplier qualification whether or not FDA does. And retrofitting 62304 documentation onto software built without it costs more than doing it from the start, because the standard is about traceable process, and process can’t be reconstructed after the fact. The recognized version is IEC 62304:2006 with Amendment 1:2015, sometimes called Edition 1.1. Edition 2 is targeted for mid-to-late 2026, and Edition 1.1 stays the recognized version until FDA updates its list.
Can a companion app be built with React Native or Flutter for a regulated device?
Yes, and the framework isn’t the thing FDA reviews. Neither FDA guidance nor IEC 62304 names an approved language or toolkit; what gets examined is your process, your risk controls, and your evidence. What cross-platform does change is your SOUP burden: every framework and plugin dependency becomes software of unknown provenance under IEC 62304, so each one needs documentation and a justification that its known defects don’t create unacceptable risk, and each one also lands in your SBOM, which for cyber devices is now a statutory requirement. The line most teams settle on is cross-platform for the interface, native for the Bluetooth layer: when a vulnerability surfaces in a platform Bluetooth stack, a native team patches it directly, while a cross-platform team waits for a plugin maintainer. If BLE reliability is the core of your product, that wait is the risk you’re accepting. We’ve built exactly this: Joovv’s companion app is React Native, BLE-connected, documented against IEC 62304, and HIPAA compliant, supporting both legacy and current hardware.
What is the difference between a companion app and SaMD?
The two terms answer different questions. Companion app describes a relationship: the software pairs with a hardware device. SaMD describes a function: software that performs a medical device function on its own, without being part of a hardware device. They overlap rather than compete, so a companion app can be a non-device display, an accessory, or full SaMD. The test that separates them: could the software perform its medical function with no paired device at all? If yes, it’s SaMD. If it supplements, augments, or controls the hardware, it’s an accessory, regulated as part of that device’s world rather than on its own. For the full standalone pathway, see our <a href=”https://topflightapps.com/ideas/samd-app-development/”>SaMD app development guide</a>.
Does the EU MDR classify companion apps differently than the FDA?
Yes, and usually more strictly. FDA sets your burden by device category and predicate, which is why a passive display can stay lightly regulated while only alerts, interpretation, or dosing commands trigger their own clearance. The EU works the other way around: under Annex VIII Rule 11 of the Medical Device Regulation, software is classified by the clinical use of the information it provides, so anything informing a diagnostic or therapeutic decision starts at Class IIa and climbs from there, with vital-sign monitoring at IIb. Accessories are classified in their own right, so pairing with a Class III implant doesn’t automatically make your app Class III, but Rule 11 will likely put it in notified-body territory anyway. The practical consequence: don’t build a US launch plan that assumes EU parity. A simplification proposal that would soften Rule 11 is pending, and current rules apply until it isn’t.