
The FDA has cleared more than 1,300 AI and machine learning-enabled medical devices. A significant number of health AI products on the market right now have not been cleared — and their founders either don’t know clearance is required or have decided to operate without it and hope enforcement doesn’t reach them. Both positions carry real risk. For any digital health startup or health tech founder, is my health AI a medical device? is a question you need to answer before launch plans, customer conversations, or Series A due diligence — not after.
The core problem: the FDA’s definition of a medical device is broad and technology-neutral. The word “AI” does not appear in the Federal Food, Drug, and Cosmetic Act. What matters is not the technology — it is what your product is intended to do and what happens if it does it wrong. A symptom checker that tells someone they probably have a cold when they have appendicitis is a medical device problem, not a search algorithm problem.
Below, you will get a five-question self-assessment that gives founders a preliminary answer, a scenario matrix covering ten common health AI product types with real named examples, and a clear action path for three likely outcomes — making this a practical starting point for product planning, health AI investment, and faster regulatory triage.
This article is not regulatory or legal advice. For a determination specific to your product, engage a regulatory affairs professional or FDA-cleared consultant.
Is my health AI a medical device? Do I need FDA clearance?
If your health AI analyzes patient data to diagnose, treat, monitor, or recommend care, it likely meets the FDA’s definition of a medical device. Whether you need clearance depends on who uses the output, whether the reasoning is reviewable, and whether an exemption — general wellness, administrative, EHR, or clinical decision support — applies to your specific product.
Key takeaways
- The FDA classifies health AI based on intended use, not the technology behind it — your product’s clinical function and marketing claims determine your regulatory status.
- Four exemption categories (updated January 2026) may cover your product without clearance, but each has specific criteria that must be documented and defended.
- Use the five-question self-assessment in this article for a preliminary verdict, then engage a regulatory affairs professional before launch for any product that touches clinical decisions.
Table of Contents
- What Makes a Health AI Product a Medical Device Under FDA’s Definition?
- The Four Exemption Categories That May Cover Your Health AI (No Clearance Needed)
- The 5-Question Self-Assessment: Does My Health AI Need FDA Clearance?
- The Health AI Scenario Matrix — 10 Common Products, FDA Classification, and Verdict
- The Three Clearance Pathways — What Actually Happens If You Need FDA Clearance
- The Gray Zone: Five Health AI Products Where FDA Classification Is Genuinely Unclear
- What to Do After the Self-Assessment — Your Three Possible Action Paths
- Why Choose Topflight Apps for Health AI Development and Regulatory Strategy
What Makes a Health AI Product a Medical Device Under FDA’s Definition?
A health AI software medical device is any software — including AI and machine learning models — that is intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease, or intended to affect the structure or function of the body. That is the statutory definition under Section 201(h) of the Federal Food, Drug, and Cosmetic Act (FD&C Act), and it applies regardless of the technology used to build it.
For health AI founders, three words do the most work: intended for use. FDA determines device status based on intended use, which includes not just what you say your product does but what your marketing claims, UI copy, sales materials, and the product’s actual clinical function collectively imply. Your indications for use — the specific condition, patient population, and clinical context your product targets — shape how FDA classifies you.
FDA applies a two-part test to software:
Part 1 — The Intended Use Test
Is the software intended to diagnose, prevent, mitigate, treat, or monitor a disease or condition? If yes, it meets the medical device definition. Internationally, this is where the IMDRF SaMD framework aligns: Software as a Medical Device (SaMD) is software that performs a medical function without being part of a hardware device.
Part 2 — The Enforcement Policy Test
Even if a product meets the device definition, FDA enforcement discretion covers several software categories — meaning they are technically medical device software (MDSW) but FDA has stated it will not enforce clearance requirements for them.
This is where the exemptions live: general wellness, administrative, EHR, and clinical decision support software. Products that do not qualify for any exemption are non-device software only if they never met the definition in Part 1.
The practical implication: meeting the device definition is not the end of the analysis. It is the beginning. The real question is whether your product falls into an exempted category or whether it requires a clearance pathway — a distinction that depends on product-specific facts, not broad labels.
If you are building AI that touches clinical workflows, understanding where AI in healthcare compliance obligations begin is essential context for this analysis.
The Four Exemption Categories That May Cover Your Health AI (No Clearance Needed)
Most health AI founders are not asking whether their product meets the medical device definition — they are asking whether an exemption covers them. Under AI medical device FDA regulation, four categories of health software meet the technical device definition but are exempt from clearance requirements.
These categories originate from FDA guidance documents issued between 2016 and 2022, with significant revisions to both the CDS and general wellness guidance issued in January 2026 that broadened several exemptions.

Exemption 1 — General Wellness Software
The general wellness exemption covers software that promotes healthy behaviors without making disease-specific claims. The test: would a reasonable person interpret this product as treating, diagnosing, or monitoring a medical condition? If no, it qualifies as a wellness app.
Qualifies: a step-counter app that encourages daily walking, a sleep hygiene app that suggests a consistent bedtime, a hydration reminder. Does not qualify: a “wellness” app that uses AI to detect irregular heartbeat patterns and presents findings in a clinical context, even if marketed as wellness. The intended use — not the label — determines classification.
The January 2026 Expansion
FDA’s updated General Wellness guidance now treats non-invasive monitoring features — such as optical sensing that estimates blood pressure, oxygen saturation, blood glucose, or heart rate variability — as general wellness products when the outputs are intended solely for wellness uses and not implied for medical or clinical purposes. This is a meaningful change for wearable and patient engagement app developers: a wrist-worn device that outputs pulse rate and blood pressure for activity and recovery tracking can qualify as general wellness, provided the product’s marketing and functionality do not imply clinical use.
The Marketing Trap Still Applies
Many founders label products “wellness” in their privacy policy while their marketing copy says “detect early signs of [condition]” or “AI-powered health screening.” FDA looks at the totality of claims. A wellness label does not override clinical marketing language.
Exemption 2 — Administrative Software
Administrative software supports operational functions in a clinical setting without directly influencing clinical decisions. Scheduling, billing, claims processing, patient communication routing, appointment reminders — all exempt. AI that optimizes appointment scheduling is not a device. AI that triages which patients need urgent clinical attention based on health data is likely a device. The line is whether the software’s output touches a clinical workflow tool that shapes care decisions or stays purely operational.
Exemption 3 — EHR and Care Coordination Software
EHR software that stores, displays, or transmits patient information — including AI features that organize, summarize, or route that information — is exempt as long as it does not make clinical recommendations. An AI that generates a structured clinical summary from free-text notes is generally exempt. An AI that reads those notes and recommends a diagnosis or treatment change is not.
This is where AI scribe tools like Nuance DAX and Abridge sit. They transcribe and structure clinical conversations under enforcement discretion as documentation tools. The moment such a tool adds a “suggested diagnosis” or “recommended next step” field, it crosses into CDS or SaMD territory. Understanding how AI will help change EHR systems means understanding exactly where this line falls.
Exemption 4 — Low-Risk Clinical Decision Support (CDS)
This is the most important and most misunderstood exemption for health AI founders. Under the 21st Century Cures Act, low-risk CDS software is exempt from FDA device regulation only if it meets all four of the following conditions:
- The software does not acquire, process, or analyze a medical image, a signal from an in vitro diagnostic device, or a pattern or signal from a signal acquisition system.
- The software displays, analyzes, or prints medical information that is generally available to healthcare professionals.
- The software is intended for use by a healthcare professional — not a patient — to support clinical judgment.
- The healthcare professional can independently review the basis of the software’s recommendation. The software shows its work.
All four conditions must be met. Fail one, and the CDS exemption does not apply.
What Changed in January 2026
FDA’s updated CDS guidance relaxed two positions that previously excluded many health AI products. First, CDS software that produces a single treatment recommendation — rather than a list of options — now falls under enforcement discretion rather than being automatically classified as a device. Second, FDA struck language that had excluded software producing a risk probability or risk score from the CDS exemption. For founders building AI risk scoring or predictive analytics tools for clinician use, this is a direct expansion of the exempt category.
The Condition Most Founders Still Fail Is The Third
If your CDS tool is used directly by patients — not by a licensed clinician who reviews the recommendation before acting — it does not qualify for the CDS exemption, regardless of how low-risk the content appears. Patient-facing AI that influences clinical decisions is almost always subject to FDA oversight.
The 5-Question Self-Assessment: Does My Health AI Need FDA Clearance?
Does my health app need FDA clearance? Work through these five questions in order. Each question gates the next — stop at the first “Yes” that changes your path.
| # | Question | If YES | If NO |
| Q1 | Does your AI analyze patient health data to support or make a diagnosis, prognosis, treatment recommendation, or monitoring decision? | You meet the device definition. Continue to Q2 to assess exemptions. | Your AI may be administrative, wellness, or EHR software. You likely do not require clearance. Confirm with Q5. |
| Q2 | Is your AI used directly by patients (not by a licensed clinician who reviews the output before acting)? | CDS exemption does NOT apply. Clearance very likely required. Proceed to Q5. | Clinician-facing AI. CDS exemption may apply. Continue to Q3. |
| Q3 | Does your AI analyze a medical image, signal from an in vitro diagnostic device, or physiological signal from a monitoring device? | CDS exemption does NOT apply (statutory exclusion). Clearance almost certainly required. | Not imaging or signal analysis. CDS exemption still possible. Continue to Q4. |
| Q4 | Can the clinician independently review the basis for your AI’s recommendation — does the software show its reasoning in a way a qualified professional could verify without relying on the AI? | CDS exemption likely applies. Confirm with Q5. Clearance probably not required. | Black-box AI output with no reviewable basis. CDS exemption does NOT apply. Clearance very likely required. |
| Q5 | If your AI malfunctions or gives a wrong output, could the result directly harm a patient — delay diagnosis, cause incorrect treatment, or miss a critical condition? | Regardless of exemption analysis, engage a regulatory affairs professional before launch. The patient safety stakes require expert review. | Lower-risk profile. If prior questions point to an exemption, you likely do not require clearance. Document your reasoning. |
This framework applies whether you are building an AI diagnostic tool, a symptom checker, a triage algorithm, predictive analytics for clinical deterioration, or a large language model (LLM) generating patient-facing responses. The underlying logic is the same: what does the software do, who uses its output, can that output be independently verified, and what happens if it is wrong?

A few worked examples to make the decision tree concrete.
A clinician-facing clinical decision support software tool that flags potential drug interactions, shows the supporting evidence, and lets the prescriber decide: Q1=Yes, Q2=No, Q3=No, Q4=Yes, Q5=depends — likely CDS-exempt.
A patient-facing conversational AI in healthcare app that triages symptoms and recommends whether to seek emergency care: Q1=Yes, Q2=Yes — clearance very likely required, regardless of how the remaining questions land.
An AI scribe that documents a clinical encounter without making recommendations: Q1=No — likely no clearance required.
This self-assessment is a preliminary screening tool, not a regulatory determination. It is based on published FDA guidance as of this article’s publication date. FDA guidance on AI/ML-based SaMD is actively evolving. A qualified regulatory affairs professional must make the final determination for any product approaching market.
The Health AI Scenario Matrix — 10 Common Products, FDA Classification, and Verdict
If you want the clearest shortcut to FDA AI health app requirements, stop focusing on whether your product “uses AI” and focus on what it is intended to do. That is the hinge. The same model can land in very different buckets depending on whether it documents, informs, triages, diagnoses, or recommends treatment. In general, patient-facing tools that influence clinical action face more scrutiny than clinician-facing software that mainly organizes information.
Here is the practical matrix:
| AI Product Type | Intended Use | Likely FDA Classification | Clearance | Real-World Example |
| AI Symptom Checker (patient-facing) | Suggests possible causes and urgency levels to patients | Likely SaMD; often Class II territory | Often yes, depending on claims and specificity | Ada-style symptom tools |
| AI Clinical Documentation / ambient AI | Transcribes visits and produces an AI-generated clinical note | Usually non-device / EHR-adjacent documentation software | No, if it stays in documentation | Abridge, Suki, Dragon Copilot |
| AI Radiology Image Reader | Detects findings on CT, X-ray, or other scans | SaMD; often Class II, sometimes higher-risk | Yes | Aidoc, Viz.ai, Paige |
| AI-Powered Triage Chatbot (patient-facing) | Routes patients based on urgency or probable condition | Likely device software; patient-facing AI weakens CDS arguments | Often yes | Symptom-based triage bots |
| Clinician-Facing Risk Scorer | Presents deterioration or sepsis-style scores with supporting factors | Potentially exempt CDS if the clinician can independently review the basis | Sometimes no | EHR-embedded risk models |
| AI Drug Interaction Checker | Flags known medication conflicts for prescribers | Often low-risk CDS / enforcement-discretion territory | Often no | Common EHR medication safety tools |
| AI Mental Health Chatbot (therapeutic claims) | Delivers CBT-like support or treatment claims to patients | Likely SaMD if positioned as treatment | Often yes, or at least serious FDA strategy needed | Woebot-style products |
| Population Health Analytics | Identifies cohorts for outreach using aggregate data | Usually non-device / administrative analytics | No | Outreach and care-management platforms |
| AI-Powered ECG Interpretation | Detects AFib or other patterns from waveform data | SaMD; commonly Class II | Yes | Apple, AliveCor |
| LLM-Based Patient Q&A / Education Tool | Answers patient questions using general health content | Often non-device if it avoids diagnostic or treatment claims | Usually no, unless personalized clinical recommendations creep in | General education bots |
A few rows deserve extra emphasis.
Radiology AI and other forms of medical image analysis AI are among the clearer cases: if software analyzes images to detect clinically meaningful findings, FDA oversight is usually part of the picture.
By contrast, ambient AI and other natural language processing tools that generate documentation usually stay in a safer lane if they remain documentation tools. That is where broader gen AI in healthcare discussions often get muddy: not every LLM feature is a device, but some clearly become one once they start driving clinical decisions.
The harder cases are clinician-facing AI risk scoring and patient-facing AI chatbot products. The key question is whether a clinician can independently review the basis for the recommendation rather than rely mainly on the software. That can support a CDS exemption for some clinician-facing tools.
Patient-facing triage and therapeutic bots have a weaker argument, especially when they steer care directly. And separately, ChatGPT HIPAA compliance is a different question altogether: privacy and security obligations are not the same thing as FDA device classification.
The basic pattern is simple: documentation, workflow, and aggregate analytics are often safer; diagnosis, image analysis, waveform interpretation, and patient-specific treatment guidance are where clearance risk climbs.
The Three Clearance Pathways — What Actually Happens If You Need FDA Clearance
If your self-assessment points to clearance, the next question is the pathway. The SaMD FDA clearance health AI founders ask about most often follows one of three routes, each with different timelines, costs, and evidence requirements.
Before committing to any of them, understand what each demands — and how the cost of AI in healthcare development compounds when regulatory preparation is underestimated.
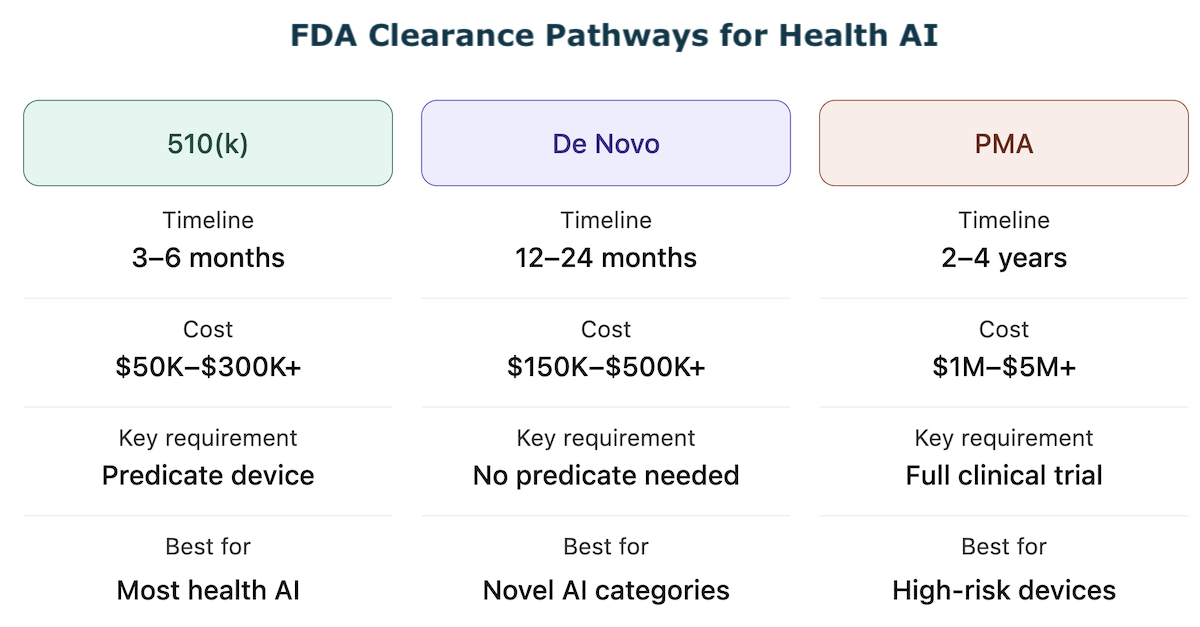
510(k) — Premarket Notification (Most Common for Health AI)
The FDA 510(k) pathway requires demonstrating substantial equivalence to a legally marketed predicate device — a previously cleared product with the same intended use and similar technological characteristics. For AI/ML health software, there are now hundreds of cleared predicates to reference, classified under specific regulations such as 21 CFR Part 880 and associated product codes.
Timeline: 3–6 months for a well-prepared submission. The 2025 median review time was approximately 142 days, though FDA questions and additional information requests can extend this significantly.
Cost: $50K–$300K+ depending on clinical study requirements, device complexity, and regulatory consultant involvement. The key requirement is identifying the right predicate device — this single decision determines whether 510(k) is available to you at all.
AI-specific consideration: FDA’s predetermined change control plan framework — finalized and now included in roughly 10% of new AI/ML clearances — allows cleared AI devices to update their algorithms within pre-agreed parameters without filing a new 510(k) submission. For adaptive algorithm products, building a PCCP into your initial 510(k) timeline is a meaningful long-term advantage.
De Novo — For Novel, Low-to-Moderate Risk AI With No Predicate
When your AI device has no clear predicate, the De Novo pathway creates a new device classification and establishes special controls for future similar devices.
Timeline: 12–24 months typically.
Cost: $150K–$500K+, requiring more extensive clinical evidence than 510(k).
The strategic upside: if granted, your device becomes the predicate for future 510(k)s in the same category. Apple’s ECG app for AFib detection went through De Novo in 2018 and is now the benchmark predicate for multiple subsequent ECG analysis clearances. Being the De Novo first-mover in a product category is a durable competitive moat and a strong signal for health AI investment conversations.
Premarket Approval — High-Risk AI, Class III Devices
PMA (Premarket Approval) is required for Class III devices where 510(k) or De Novo is not available — typically devices that sustain or support life or present a potential unreasonable risk of harm. Most software-only AI health products do not reach Class III, but AI that interprets cardiac images for life-threatening conditions or controls implantable device parameters may.
Timeline: 2–4 years.
Cost: $1M–$5M+, with full clinical trial data required. Relevance to most health AI founders: low — escalate to regulatory counsel if Class III is indicated.
The Pre-Submission (Q-Sub) Meeting — Use This Before Committing to a Pathway
Before investing in any clearance pathway, schedule a pre-submission meeting with FDA’s Digital Health Center of Excellence. This is a free, non-binding FDA pre-submission consultation where you describe your product and receive informal feedback on classification and pathway. It does not delay your 510(k) timeline — it prevents you from spending six months on a 510(k) when De Novo was required, or filing a 510(k) when enforcement discretion actually applies.
The Gray Zone — Five Health AI Products Where FDA Classification Is Genuinely Unclear
Not every health AI product falls neatly into “device” or “not a device.” Some sit in the regulatory murk where intended use, workflow design, and the level of human review matter more than the model itself.
This is where both HIPAA and FDA questions tend to collide, and where teams shipping fast with HIPAA compliance for AI-generated code can accidentally build themselves into a riskier classification than they intended.
1. LLM-Based Clinical Summarization With Personalized Recommendations
An LLM that summarizes a chart for a clinician usually looks like non-device EHR software. But if it adds, “consider ordering an HbA1c based on this history,” it starts to look like CDS. The key question is whether the clinician can independently review the basis for that recommendation or is expected to trust the model.
2. AI-Powered Prior Authorization Tools
If the AI predicts approval odds or auto-fills forms, that is generally administrative software. If it starts recommending whether a treatment is clinically appropriate and that output influences care decisions, it can drift toward CDS territory. That is a very different regulatory animal.
3. Remote Patient Monitoring AI That Generates Alerts
Displaying raw vitals is usually non-device software. An RPM tool that analyzes trends and pushes alerts like “review medication titration” sits right on the boundary. If a clinician reviews the underlying data, it may remain exempt; if it triggers action automatically, device risk increases.
4. AI Nutrition and Metabolic Health Apps With Clinical Claims
A calorie tracker is wellness software. An app that combines CGM, food logs, and labs to infer early insulin resistance is making a diagnostic-style claim. That mix of sensor data, inference, and clinical conclusion is exactly where a locked algorithm can start looking like regulated software, and an adaptive algorithm can raise even harder questions.
5. Generative AI for Patient-Facing Mental Health Support
This is one of the fastest-moving corners of generative AI healthcare. Low-intensity support may fall under enforcement discretion, but once a tool evaluates suicide risk or adjusts interventions, scrutiny rises fast. Founders here should watch FDA closely and also think beyond marketing claims; therapy chatbot compliance is not just a privacy issue. Even adjacent tools such as population health analytics can become riskier once they start shaping patient-level action.
What to Do After the Self-Assessment — Your Three Possible Action Paths
After working through the five-question framework, you are in one of three situations.
Path A — Clearance Not Required (Exemption Applies)
Document your reasoning. Write down which exemption applies and why — general wellness, administrative, EHR, or CDS — with reference to the specific FDA guidance document. This documentation is your defense if FDA ever questions your classification. Review annually as your product evolves — adding a new AI feature can change your classification.
Specifically:
- document the exemption category with its regulatory basis,
- write an intended use statement reviewed against exemption criteria,
- audit your marketing copy for language that contradicts your exemption claim,
- schedule an annual review tied to your product roadmap.
If your product also handles protected health information, HIPAA compliant app development requirements apply independently of your FDA status.
Path B — Clearance Required
Step 1: Schedule a Q-Sub meeting with FDA’s Digital Health Center of Excellence — free, non-binding, high-value.
Step 2: Engage a regulatory affairs consultant to confirm classification and identify the clearest predicate device for a 510(k).
Step 3: Do not market the product as a diagnostic or therapeutic tool until clearance is obtained.
Step 4: Begin QMS implementation — clearance submission requires ISO 13485-aligned development documentation.
Each step protects your time to market and reduces rework.
Path C — Gray Zone
Engage a regulatory affairs professional before launch. The cost of a proper regulatory opinion ($5K–$20K) is a fraction of the cost of an FDA enforcement action, a forced product recall, or failed due diligence on a Series A. The Q-Sub meeting is free and non-binding — use it.
Operating in the gray zone without documentation is not a neutral act. If FDA reviews your product and finds no evidence you considered the regulatory question, that is an aggravating factor. Documented good-faith analysis is a mitigating factor. Write it down either way.
Why Choose Topflight Apps for Health AI Development and Regulatory Strategy
Topflight builds health AI products across the clearance spectrum — from HIPAA-compliant wellness apps that are clearly non-device to FDA 510(k)-ready clinical decision support tools. We understand both the technical build and the regulatory documentation that health AI FDA clearance requires. What we deliver:
- Regulatory classification assessment — mapping your AI feature set to the correct FDA tier before development begins
- FDA-ready development documentation — QMS-aligned process, intended use documentation, risk management files
- HIPAA-compliant architecture — encryption, audit logging, BAA management
- 510(k) and De Novo preparation support — software documentation package for FDA submission
- Existing AI-built product audits — identifying regulatory gaps in products already in market
The FDA has cleared more than 1,300 AI/ML medical devices. The founders who navigated AI health app FDA approval — even when it was expensive and slow — have defensible products, investor credibility, and clinical customers that the founders who skipped it cannot reach. Know where your product stands. We can help you get there.
Frequently Asked Questions
Does my health app need FDA approval?
It depends on intended use. If your app uses AI to diagnose, treat, monitor, or recommend care for a medical condition, it likely meets the FDA’s medical device definition. Apps that are purely wellness, administrative, or EHR documentation tools generally do not require clearance.
What is the difference between a wellness app and a medical device?
A wellness app promotes healthy behaviors without making disease-specific claims — step counting, sleep hygiene, hydration reminders. The moment an app uses AI to detect, diagnose, or monitor a medical condition, it crosses into medical device territory regardless of how it is labeled.
Is symptom checker a medical device?
In most cases, yes. A patient-facing AI that analyzes symptoms and suggests possible diagnoses or triage decisions meets the FDA’s device definition and does not qualify for the CDS exemption because it is not clinician-facing.
Does a clinical decision support tool need FDA clearance?
Not if it meets all four criteria under the 21st Century Cures Act: it does not analyze images or physiological signals, displays generally available information, is intended for clinician use, and shows reviewable reasoning. Fail any one and clearance applies.
How long does it take to get FDA clearance for a health AI product?
For 510(k), the median review time is approximately 4–5 months. De Novo takes 12–24 months. PMA requires 2–4 years. Preparation time before submission adds months to each pathway.