
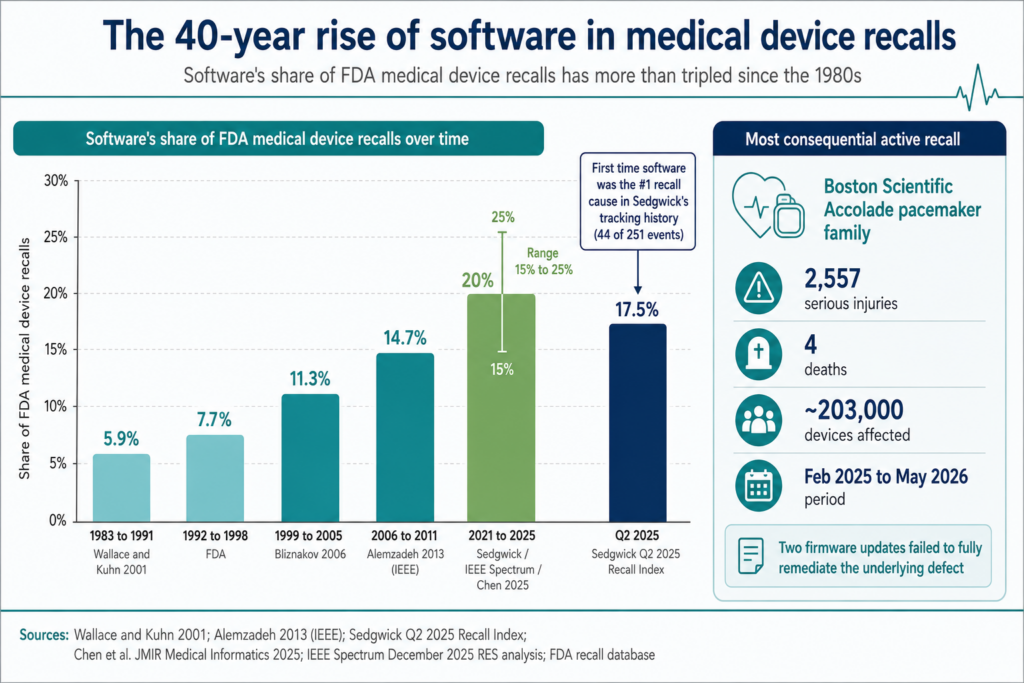
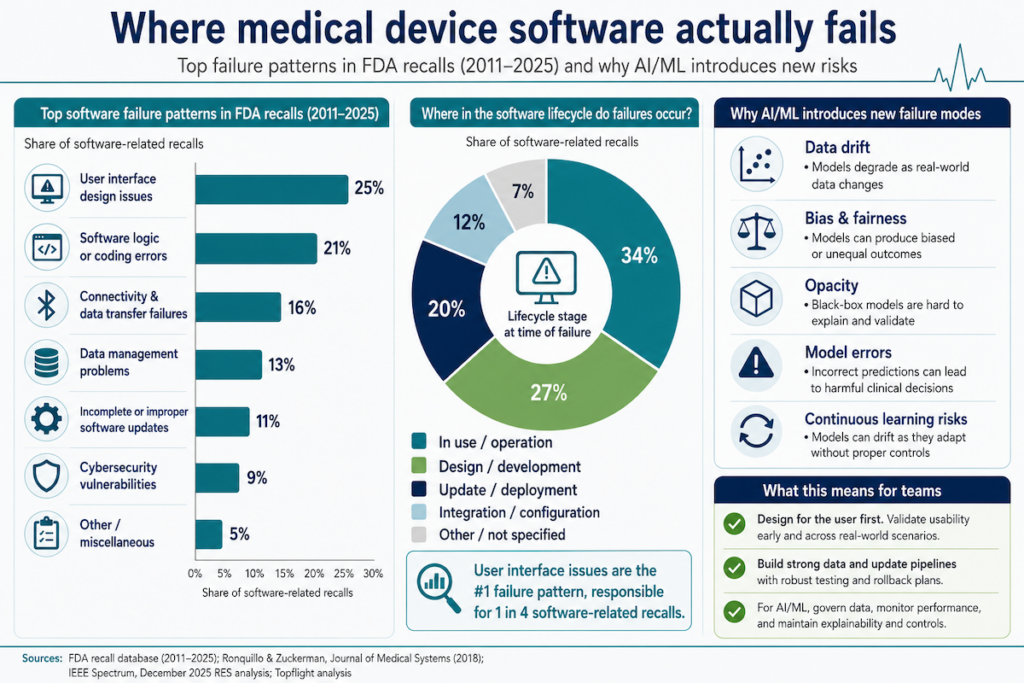
In medical device app development, software is now the single largest cause of FDA recalls. In Q2 2025, software root causes drove 44 of 251 recall events tracked by Sedgwick. That’s the first time software displaced device failure as the top category in the index’s history.
The trajectory took four decades to get here. Software accounted for 5.9% of recalls in the 1980s, 7.7% through the late 1990s, 11.3% by 2005, 14.7% by 2011, and somewhere between 15% and 25% across 2021 to 2025. In 2024, Tandem Diabetes Care recalled its t:connect iOS app after a crash loop triggered Bluetooth flooding that drained t:slim X2 pump batteries and stopped insulin delivery. 224 injuries in the first tally.
Boston Scientific’s Accolade pacemaker recall has logged 2,557 serious injuries and 4 deaths across ~203,000 devices since February 2025. Two firmware updates failed to fully remediate the underlying defect.
When medical device software fails, it fails in the software layer first. This piece covers what we’ve learned from developing medical device software that survives FDA review and ships without ending up in a recall notice. If you’re working out how to develop software as a medical device without joining the index, the sections below walk through what holds up under audit.
Top Takeaways:
- Effective medical device software and app development hinges on matching your build to current regulatory standards. Treat regulation as architecture, baked in from the first sprint.
- The medical device software development process runs from concept to deployment. Errors caught late, in validation or post-market, are the ones that produce recalls.
- Software development for medical devices increasingly means stitching new features into legacy hospital systems. Plan integration work as first-class scope.
- AI and IoT extend what’s possible in remote patient monitoring and clinical analytics. They also bring fresh regulatory questions about how machine outputs influence care decisions.
Table of Contents:
1. Understanding medical device software
2. Types of medical device software
3. Key stakeholders in medical device software development
4. Benefits of medical device software development for healthcare businesses
5. Challenges in medical device software development
6. Comprehensive medical device software development lifecycle
- Step 1: Concept and feasibility
- Step 2: Requirements and specifications
- Step 3: Design and architecture
- Step 4: Development and implementation
- Step 5: Testing and validation
- Step 6: Deployment and maintenance
7. Medical device software development standards and certifications
8. Emerging technologies in medical device software development
9. Key performance indicators for medical device software projects
10. Cost considerations for medical device software projects
11. Security considerations in medical device software development
12. Future trends in medical device software development
13. How Topflight helps you build medical device software
Understanding medical device software
Medical device software development covers more than SaMD. IoMT, embedded systems, clinical information platforms, and the integration glue between them each carry different growth curves and different regulatory weight.
Medical device SaMD sits at one end of the spectrum, embedded software inside regulated hardware sits at the other, and SaMD software for clinician workflows fills the middle.
SaMD is growing more than 2x faster than the broader medical device market
The US Software as a Medical Device (SaMD) market is forecast to grow at a 13.5% CAGR through 2034. The broader US medical devices market grows slower, at 5.28% CAGR from 2024 to 2029, reaching $232.6 billion by 2029. Globally, SaMD tracks similarly at 13% CAGR through 2034, hitting $6.1 billion. North America holds 23.9% of that market share in 2024.
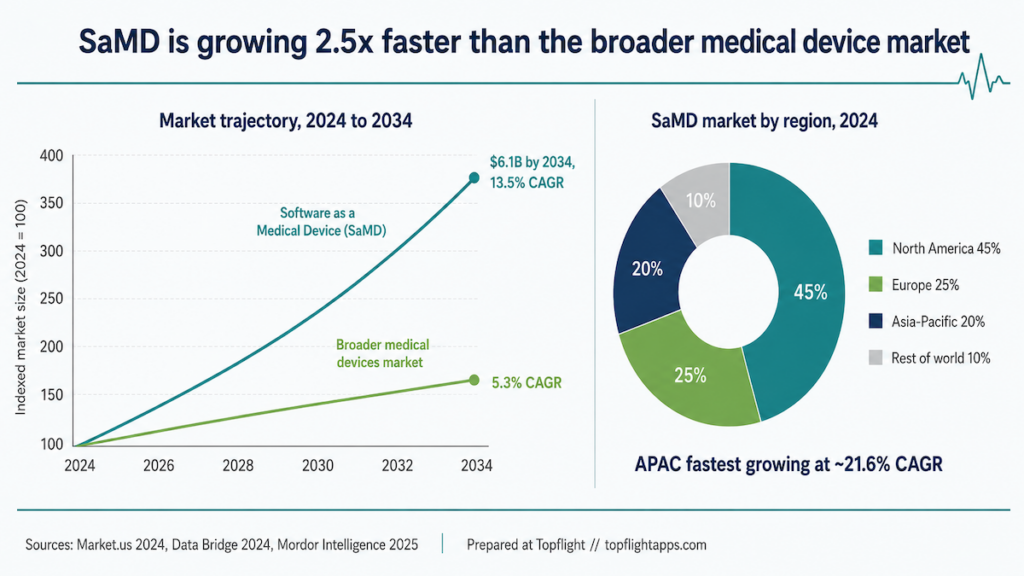
Two demand-side drivers explain the curve: more digital health tools getting adopted into clinical workflows, and a rising chronic-disease patient base that wants continuous patient monitoring.
If your build pulls in EHR data, see our notes on EHR data migration for the timeline traps we keep seeing.
Types of medical device software
Four categories matter, split along one axis: software that is the device, and software that orbits it.
Software as a Medical Device (SaMD) is the device
SaMD is software that does medical work without being part of any hardware device. Mobile medical apps and clinical decision support tools are the most common examples. It runs on smartphones, in browsers, on cloud platforms, or anywhere else general-purpose compute is available.
If your SaMD will hand recommendations back to clinicians, see our notes on clinical decision support system implementation for the integration timeline and audit-trail patterns that hold up under review.
Internet of Medical Things (IoMT): the connective tissue
IoMT is the layer of interconnected devices in healthcare environments that collect and transmit health data. Smart wearables and remote patient monitoring tools are the obvious cases. The continuous monitoring and real-time data they generate give clinicians a view of what’s happening between visits.
Embedded medical software ships welded to the hardware
Embedded medical software is the code living inside pacemakers, infusion pumps, imaging machines, and ventilators. It controls device function and has to operate safely under failure conditions you can’t recover from with a reboot.
Clinical information management platforms: the layer everything else plugs into
This is the layer of electronic health records (EHR), hospital management systems, laboratory information systems, and revenue cycle platforms that everything else has to integrate with. The integration into these clinical workflows is where most medical device software timelines slip.
Key stakeholders in medical device software development
On any medical device software build, five stakeholder groups have skin in the game and their non-negotiables differ. Treating them as one audience is how projects ship a clean V1 that nobody adopts.
Healthcare providers: zero patient harm in 30-second clinician windows
Hospitals and clinics need software that handles clinical decision-making and patient throughput without making clinician work harder. Non-negotiables:
- Safety and time: Zero patient harm, tasks that fit a 30-second clinician window, alarm-fatigue controls, downtime modes, and graceful degradation when networks drop.
- Interoperability: Native FHIR R4/HL7 v2, Epic/Cerner-friendly auth (OIDC/SAML SSO), and bulletproof patient/encounter matching.
- Security and compliance: BAA, PHI minimization, audit trails, role-based access, MDM for the fleet, cert pinning, and SBOM with a patch plan.
- Operations: Versioned configurations, safe rollback, environment parity, and SLAs that name response times and uptime. The on-call path needs to be one you can actually reach.
- Proof it works: Adoption curve, alert-to-action conversion, task time reduction, and outcome deltas (readmission, LOS, no-show rate). If you can’t measure it, it didn’t happen.
Medical device manufacturers: build for submissions and the field
Companies that build the devices themselves need software that survives both regulatory submission and field conditions. Build for submissions, and build for the field.
- Regulatory backbone: IEC 62304, ISO 14971, IEC 62366-1, and ISO 13485, with full traceability (hazard → requirement → test → evidence) and clean DHF/DMR.
- Architecture: Safety partitions, watchdogs, secure boot, signed OTA, and forensics-grade logs that don’t leak PHI.
- V&V depth: Hardware-in-the-loop, worst-case timing, soak tests, and failure-mode drills (power loss, radio drop, bad firmware, sensor failure).
- Manufacturing and service: Provisioning, calibration, golden image management, and remote diagnostics that won’t brick devices.
Use one reusable platform across SKUs, with modular features gated by configuration. Forking per SKU is the failure mode. Pre-Sub meetings de-risk the 510(k)/De Novo story.
Patients: if they won’t use it, nothing else matters
Patients use the result. Adoption hinges on usability that meets WCAG 2.2 AA, with:
- plain language,
- large tap targets,
- multilingual support,
- offline-first design,
- caregiver access for the people doing the actual care.
The safety net matters as much as the happy path: clear “what now?” guidance on abnormal readings, human-in-the-loop where appropriate, time-to-response guarantees, and fallback paths that hold when connectivity drops.
Privacy lives by default. Plain-language consent, granular sharing controls, export and delete options, and edge processing where feasible. No dark patterns. Ever.
Engagement nudges should be calibrated to behavior, with streaks only when they help adherence and reminders sized to the actual risk. Apple Health and Google Fit integrations reduce manual effort, which matters for people whose chronic conditions already eat their day.
Equity stays in scope: low-bandwidth mode, device-agnostic support, UX validated on hardware three generations old, and accessibility specs that survive an actual screen reader.
Regulators: evidence packages that survive inspection
Regulators (FDA, EMA, IRBs, EU notified bodies) need software that produces evidence they can act on. Make auditors and IRBs smile, or at least stop frowning.
- Regulatory packages: Crisp intended use, SOUP inventory, cybersecurity posture (threat model, SBOM, coordinated disclosure, incident response runbook), patch cadence, and post-market surveillance plan.
- Evidence you can defend: Prespecified statistical analysis, ground-truth labeling audits for ML, bias checks, and versioned models with change control and rollback criteria.
- Research workflows: eConsent, IRB documentation, data provenance, HIPAA Safe Harbor/Expert Determination de-identification, and reproducible pipelines.
FHIR for clinical, OMOP for research. No bespoke schemas unless there’s a life-or-death reason. Audit-ready ops mean immutable logs and time-sync across systems. Requirement management gets mapped to:
- IEC 62304
- ISO 14971
- IEC 82304-1
- ISO 13485
That mapping keeps inspections from becoming archaeology.
Clinical researchers: trial integrity and speed by design
Researchers run trials on top of the software. The data has to survive both a statistical analysis plan and a regulatory audit. Where the bar sits:
- GCP and Part 11 by default: Time-stamped audit trails, validated e-signatures, role-based controls, and immutable provenance.
- Audit-survivable data capture and outputs: eSource↔EDC (REDCap/Medidata) interoperability, ePRO/eCOA with version-locked instruments, device telemetry with clock sync and chain-of-custody, CDISC SDTM/ADaM exports, and schema validation gates before database lock.
Randomization runs through IWRS with allocation concealment in tamper-evident logs. Unblind paths stay gated and audited.
ML endpoints follow the same discipline. Pre-register the metrics, lock the train/validate splits, monitor for site and shift drift, and ship every model version with rollback criteria.
- Monitoring and safety: DSMB dashboards, AE/SAE capture with MedDRA coding, protocol deviation tracking, and interim analyses with stopping rules.
- Privacy and ops at scale: eConsent with comprehension checks, HIPAA Safe Harbor/Expert Determination de-identification, BAA chains documented end to end, multi-site onboarding, and enrollment/retention/query KPIs.
Decision test: Could we recreate every figure in the SAP with one command?
Benefits of medical device software development for healthcare businesses
Medical device software developers deliver four kinds of benefits to the healthcare industry when the work gets done well: clinical, operational, competitive, and regulatory.
Medical device software closes the gap between data and clinical decisions
Software that lives inside the clinical workflow shortens the path from observation to action. The goal: safer, earlier interventions and fewer avoidable errors.
- Better patient care and outcomes
- Real-time monitoring and data analysis
- Personalized treatment plans based on patient data
- Earlier detection of clinical deterioration
The compound effect is evidence-based care that adapts to the patient in front of the clinician.
Healthcare device software shows up as fewer clicks and cleaner handoffs
Operational wins show up as fewer clicks, cleaner handoffs, less duplicate data entry, and faster query resolution across teams and systems.
- Improved operational efficiency
- Cleaner workflows and decision making
- Automated processes reducing manual errors
See our notes on how to integrate health app with Epic EHR EMR for the interoperability patterns and patient data flows that hold up in production.
When data lands where the work happens, throughput improves and rework drops. The team spends more time on care.
Medical software builds a competitive moat through compounding features
Software is where the compounding happens. Features stack across versions, and the gap between vendors becomes the gap between codebases.
- Faster product iteration
- Adoption of AI and IoT capabilities
- Integration depth that competitors can’t shortcut
- Switching costs that grow with every deployed workflow
Teams that build this way iterate faster and sustain defensible moats as features compound.
Regulatory compliance shortens audits and speeds partner approvals
Compliance done well reduces risk and builds trust with patients, providers, regulators, and the payors who control reimbursement. It also shortens audits and speeds partner approvals.
- Improved risk management and traceability
- Regulatory compliance posture that holds up across audits
Think secure-by-design architectures, traceable changes, validation plans that match device risk, and auditable governance. Compliance programs that work minimize surprises and smooth market access. The team gets to focus on shipping.
Challenges in medical device software development
Six categories of challenges show up consistently in medical device software development. Most projects underestimate at least one.
Legacy systems are usually the first wall medical device software hits
EHR installs, lab information systems, imaging archives, and pharmacy automation systems often lack the API surface needed to absorb new medical software without re-plumbing. The cost shows up as schedule slippage and integration debt that compounds across sprints.
Plan adapters and data mapping in the first sprint, and scope staged cutovers into the budget. Surprise refactors happen when undocumented interfaces surface, so leave room.
Multi-stakeholder design: each role gets its own view
Doctors, nurses, administrators, and IT each have different jobs to do inside the software systems. Custom medical device software designed for one role usually loses the others.
A workable approach: separate clinical, operational, admin, and IT views with tight role-based permissions, then validate workflows with real users before scaling.
Handoffs are where the medical device software development process loses time
Handoffs between teams compound. Each one adds delay and creates room for context loss. The cost shows up at the validation stage when nobody owns the integration story.
Tactically:
- shrink handoffs with cross-functional squads
- automate CI/CD with quality gates that include compliance checks
Traceability across requirements, code, tests, and validation evidence keeps velocity without losing the audit trail.
UX is the adoption gate for medical device software
User-centric design is the difference between a deployed product and one that sits unused. Clunky interfaces and bad navigation drop adoption rates for medical device development software, and adoption is what the build’s economics depend on.
See our notes on healthcare mobile app design for the patterns that drive patient engagement and hold up in clinical use.
Anchor UX to real clinical moments. Priority workflows have to work in low-light conditions and with gloves on. Error states need the same design attention as happy paths. Healthcare device software development that ignores the clinician’s actual workflow ships into a void.
Developing software in this space means writing code that has to defend itself in an audit, not just pass a test suite.
Architect for scale and interoperability before pilot success forces it
Healthcare organizations hit a wall with their healthcare software once pilots succeed and volume arrives. Existing systems often can’t handle the throughput, and integration with other medical device software engineering efforts becomes the bottleneck at scale.
Also read: A Complete Guide to Medical Device Integration
Plan for scale with:
- event-driven architectures,
- well-defined APIs,
- versioned data contracts,
- contract testing in CI.
Interoperability is a product surface that needs the same attention as user-facing features.
Regulatory compliance needs one roadmap across all jurisdictions
Medical device teams hit the trade-off between shipping speed and regulatory requirements. The teams that get this right plan compliance evidence in parallel with the build.
FDA standards are non-negotiable for any healthcare device software developer shipping in the US market. The work is risk management and traceability, with evidence packages that hold up in 510(k) or De Novo submissions.
HIPAA adds a continuous data-protection lens whenever patient data is involved. The work is:
- encryption
- access controls
- audit logging
- breach-detection tooling that survives an OCR review
Related: HIPAA Compliant App Development
Markets outside the US bring their own regimes: EU MDR/IVDR, Health Canada, TGA in Australia, and country-specific cybersecurity standards. The compliance roadmap has to cover the markets you’re shipping into, with evidence reusable across them where possible.
Use a compliance roadmap (FDA + MDR/IVDR + cybersecurity standards + post-market surveillance) to phase evidence generation and avoid last-minute audit scrambles.
The right healthcare app developer turns these challenges into planned scope. Surprise refactors are what you get when the team doesn’t have the muscle memory.
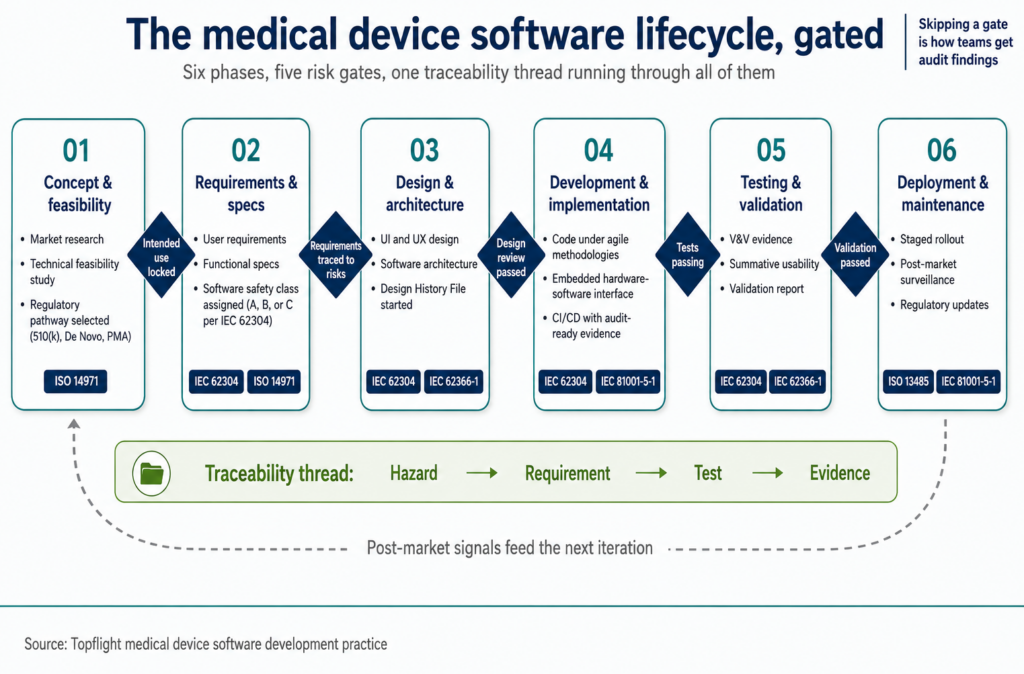
Comprehensive medical device software development lifecycle
Topflight has been a medical device research and development software developer for over a decade. The software development process for medical devices below is what we’ve refined across those builds. A medical device software developer needs all four moves:
- agile under regulation,
- QMS integration,
- risk-based development,
- and the lifecycle phases themselves.
Agile for medical devices: short cycles plus evidence at every sprint review
For regulated products, agile only works if you iterate with evidence. Use short cycles to de-risk assumptions early, and map every story and increment to design controls and validation. Keep the definition of done tied to testable requirements and traceability.
- Time-box discovery and verification tasks alongside features.
- Map each user story to requirements, risks, tests, and post-market signals.
- In sprint reviews, demo working software and updated evidence.
A story is done only when code, tests, documentation and evidence deltas, and risk file entries are all updated.
The QMS is the lane medical device software ships in
A lifecycle succeeds only when it plugs into your QMS (ISO 13485 / 21 CFR 820). Treat DHF/DMR/DHR/RMF as living artifacts; requirements, risk files, verification, and validation plans should evolve with the backlog.
- PR checklists tied to design controls and cybersecurity.
- Auditable CI that stores test results, reports, SBOMs, and signed build artifacts.
- Automatic generation of release-ready evidence bundles each increment.
- Versioned compliance evidence packages exportable per release.
Build QMS hooks into everyday delivery. Audit-readiness becomes a side effect of doing the work.
Risk-based development: ISO 14971 priority sets design depth
Lead with ISO 14971 risk thinking. The four-step rhythm:
- identify hazards
- estimate risk
- specify controls
Risk priority drives design depth, test coverage, usability work, and cybersecurity hardening. Feature size and stakeholder enthusiasm don’t outrank it. Maintain a visible map (hazard → requirement → test → evidence), scale V&V intensity with clinical risk class, treat any risk without a passing traceable test as unmitigated, and document residual risk explicitly with a sign-off path for accepting it.
Six phases of the medical device software development lifecycle
The software development process for medical devices runs through six phases. Each phase has its own deliverables and exit criteria. Skipping the criteria is how teams get audit findings.
| Phase | Key activities |
|---|---|
| 1. Concept and feasibility | Market research to identify gaps in healthcare needs. Technical feasibility study to assess development constraints. |
| 2. Requirements and specifications | Document user requirements for end-user needs. Define functional specs covering medical data handling, integration with existing systems, compliance, and post-market obligations. |
| 3. Design and architecture | Create software design covering UI and UX. Develop software architecture: structure and technologies. |
| 4. Development and implementation | Write software code, usually under agile methodologies. Handle embedded medical development for hardware-software integration. |
| 5. Testing and validation | Run unit, integration, system, and acceptance tests for quality assurance. Validate software for regulatory compliance and real-world performance. |
| 6. Deployment and maintenance | Deploy to end-users with a staged rollout. Provide maintenance through updates, bug fixes, regulatory adaptations, and post-market surveillance feedback. |
Step 1: Concept and feasibility
The concept phase covers brainstorming, technical feasibility checks, market-gap mapping, and picking the regulatory pathway you’ll target. Market research surfaces what healthcare organizations actually need and where current medical software falls short.
A technical feasibility study tests whether the proposed solution fits inside existing technological constraints. Everything downstream rests on these decisions.
Read more on healthcare app development
Step 2: Requirements and specifications
Once the concept is validated, requirements get gathered and documented in detail. The output is something stakeholders can agree on and engineering can build against.
- User requirements: Documenting what end-users need from the software.
- Functional specifications: Defining the software’s functionality, including how it handles medical data, integrates with existing medical systems, ensures compliance with regulations, and surfaces errors to clinicians.
Step 3: Design and architecture
The design and architecture decisions made here lock in the structure that engineering builds against. Software design covers the user interface, user experience, interaction flow, and accessibility behavior. Software architecture defines the underlying structure and technologies.
Step 4: Development and implementation
Coding happens here. The medical device software developer team builds against the specs and design from the previous phases.
- Coding: Writing software code, usually under agile methodologies for iterative progress.
- Embedded medical development: For devices with embedded systems, programming the hardware-software interface.
- Integration work: Wiring the new software into the EHR, device, and cloud surfaces it depends on.
Step 5: Testing and validation
Testing covers four levels:
- unit
- integration
- system
- acceptance
The point is to catch defects before the software ships and prove it meets all requirements. Quality assurance runs testing protocols that catch issues before they reach validation. Validation proves the software complies with regulatory requirements and performs in real-world scenarios.
Step 6: Deployment and maintenance
Once testing and validation pass, the software goes to production. Post-deployment maintenance addresses issues, ships updates, keeps the software current with regulations, and feeds post-market surveillance signals back into the backlog.
- Deployment: Rolling out the software to end-users in staged waves.
- Software maintenance: Regular updates and patches to address bugs, improve performance, adapt to new regulations, and incorporate post-market surveillance findings.
For where the decentralized side of this is heading, see our notes on web3 development in healthcare.

Best practices for developing medical device software start with process discipline
Process discipline shows up in how you run sprints, how you treat risk, how you involve real users, and how you document evidence as you go. The first three are where most medical device software developer teams improve fastest.
Agile methodologies work under regulation when every story carries evidence
Agile lets teams adapt to evolving requirements through short, iterative cycles. The fit is especially good in healthcare software development, where regulation, clinical workflow, patient needs, and integration scope all shift across the project.
Sprint planning breaks work into manageable chunks with clear deliverables per sprint. CI/CD automation catches issues early and ships updates faster, which matters when each release has to clear traceability before merge.
Risk management as a continuous practice
Risk management is the work itself in medical software development. Identify hazards, assess their probability and severity, mitigate the ones above threshold, and document residual risk explicitly.
- Risk assessment: Identifying potential risks at each stage of the development lifecycle.
- Mitigation strategies: Developing plans to address identified risks before they derail the project.
- Residual risk documentation: Recording what risks remain after controls, with a sign-off path for accepting them.
User-centric design with real clinicians in the loop
Real users in the loop is non-negotiable. The product meets actual needs when end-users have shaped it through feedback and usability testing throughout the build.
- User feedback: Regularly collecting and incorporating feedback from end-users.
- Usability testing: Running tests to verify the software works under real conditions and is efficient for users.
Clinical staff using the software in production conditions surface defects that lab testing never catches. The teams that win build that feedback pipeline before V1 ships.
See our breakdown of the best telemedicine apps in current production use.
Audit-ready documentation built into every sprint
Documentation written alongside the work is the only documentation that survives audit. Teams that batch it for the end produce DHFs and DMRs that don’t match the code, and the rework lands during the worst possible week.
Living artifacts get updated each sprint:
- requirements
- risk files
- V&V plans
- design history
Evidence in the PR ties test results, traceability links, and risk-file deltas to the same change that introduced the feature. Auditors find what they need without archaeology.
Quality and compliance as continuous practice across the lifecycle
Quality and compliance in medical device software development run on four habits: regulatory adherence, QA discipline, continuous improvement after launch, and audit-ready documentation throughout.
Regulatory compliance: FDA and HIPAA as concurrent threads
Compliance with FDA guidelines and HIPAA isn’t optional. Non-compliance shows up as penalties, patient harm, shipping delays that don’t recover, and partnership opportunities you don’t get.
- Regulatory guidelines: Adhering to FDA regulations, including Design Controls and Software Validation.
- HIPAA compliance: Implementing data protection measures that keep patient data private and audit logs defensible.
Quality assurance: automated and manual testing in parallel
Quality assurance has to catch defects at every level, from unit tests to acceptance tests. The output is software that’s reliable, safe, performs to spec, and ships with evidence backing each claim.
Automated and manual testing methods work in parallel:
- automated catches regression and load issues
- manual catches exploratory bugs and clinical workflow edge cases
Records of the development process, testing results, compliance checks, and design decision rationales travel with the code, so the audit trail is always one query away.
Continuous improvement after launch keeps software relevant
Software ships and immediately starts aging. Post-deployment monitoring, user feedback, regulatory tracking, and field-incident reports all feed the next iteration.
- Monitoring: Regularly tracking software performance and user feedback in production.
- Updates: Timely patches to address issues, improve functionality, comply with new regulations, and incorporate field-incident learnings.
- Regulatory tracking: Watching for updates to FDA guidance, EU MDR, and adjacent standards that affect your release planning.
Audit-ready documentation as the discipline that makes the other three defensible
The three habits above only hold up if the documentation backs them. Build the audit trail as part of the work itself, with the evidence regulators and partners actually read.
Two practical moves:
- maintain traceability from hazard through requirement and test to evidence (kept current per release)
- keep design history versioned with signed build artifacts and per-release evidence bundles
The software development process for medical devices is complex by design. Teams that ship well treat the structured lifecycle, best practices, quality habits, and risk discipline as one system. Medical device software developers who do all four deliver builds that don’t end up in recall notices.
That’s what separates teams that survive surprise inspections from teams that scramble through them.
For 24/7 patient interaction at scale, see our take on chatbots in healthcare.
Medical device software development standards and certifications
Treat regulation as architecture for medical device software. These frameworks define what evidence has to exist and how it maps to risk. Build the team, pipelines, artifacts, and review cadence to produce that evidence by default.
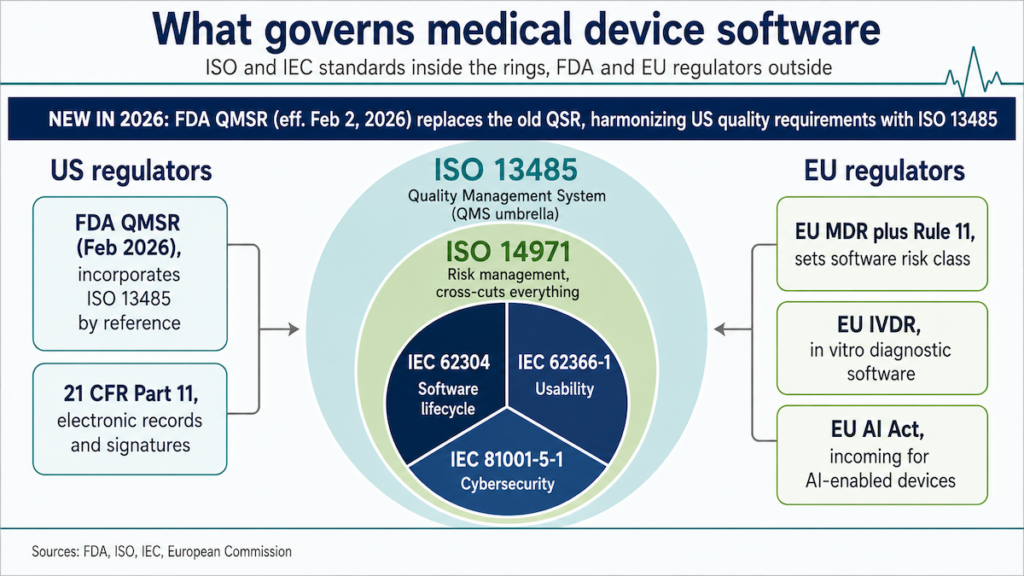
ISO 13485: the QMS for the whole build
As of February 2, 2026, FDA QMSR replaces the old QSR and incorporates ISO 13485 by reference, harmonizing US quality requirements with the international standard.
What it governs: Your QMS, covering how you plan, build, verify, release, and improve devices and their software.
Applies to: Organizations making or supporting medical devices and medical software.
Core artifacts to expect:
- Documented design controls (plans, reviews, verification/validation, release approvals), with DHF/DMR/DHR/RMF kept in sync.
- Supplier qualification plus SOUP/vendor controls tied to risk.
- CAPA with effectiveness checks; internal audits and management reviews.
- Risk file linked to design records, updated as architecture evolves.
Practical notes:
- Build QMS hooks into day-to-day delivery (PR checklists, CI evidence drops).
- Avoid the “paper QMS”: if engineers don’t touch it, auditors will.
- Tie QMS roles to code ownership so changes automatically trigger the right records.
- Make compliance evidence regenerable from source on demand.
Decision test: Could a brand-new engineer ship a change and have all required QMS evidence appear automatically?
IEC 62304 governs the medical device software lifecycle
What it governs: The software lifecycle, covering planning, requirements, architecture, implementation, verification, release, problem resolution, and maintenance.
Applies to: SaMD and SiMD (software in a medical device), with safety classes A through C driving rigor; class C carries the highest risk and the heaviest evidence load.
Core artifacts to expect:
- Software development plan plus configuration and problem-resolution procedures.
- Requirements ↔ architecture ↔ implementation ↔ verification traceability.
- SOUP inventory with risk controls; maintenance plus anomaly handling.
- Defined release process with versioned evidence packages signed at each milestone.
Practical notes:
- Set software safety class early; it dictates V&V depth and documentation.
- Keep architecture safety partitions explicit; test worst-case timing and failure modes.
- Map 62304 activities to ISO 14971 hazards/controls and IEC 62366-1 usability where risk is user-driven.
- Document SOUP decisions and their risk justifications at each acceptance.
Decision test: For each hazard, can you point to a requirement, a control, a passing test, and a residual-risk record?
FDA software validation: prove the software works for its intended use
What it governs: Under 21 CFR 820 (Design Controls) and FDA’s Software Validation guidance, you must validate software for its intended use. For electronic records and signatures, 21 CFR Part 11 may apply.
Applies to: Devices and SaMD subject to 21 CFR 820 Design Controls, and any software handling regulated electronic records or e-signatures (Part 11).
Core artifacts to expect:
- Validation plan tied to intended use; acceptance criteria pre-specified.
- Requirements and risks traceability into verification and validation activities.
- Evidence that production-equivalent builds, data, users, and environment configurations were represented.
- For Part 11: audit trails, e-sig controls, security, and record retention.
Practical notes:
- Think “fit for intended use”: simulate real users, environments, data, and workflow constraints.
- Calibrate CSV effort by risk; use automated, auditable CI to cut busywork while preserving evidence.
- Pre-Subs can de-risk your approach for novel tech.
- Document the validation rationale at the level a reviewer can follow without backstory.
Decision test: Could an FDA reviewer follow your validation logic without a meeting?
EU MDR: Rule 11 classification determines the conformity route
What it governs: Classification rules (notably Rule 11) determine the risk class. The class drives the conformity route and the evidence burden for software under EU MDR.
Applies to: Software with a medical purpose placed on the EU market (including SaMD).
Core artifacts to expect:
- Classification rationale (e.g., Rule 11), intended purpose, risk class, and conformity route selected.
- Technical documentation: GSPRs mapping, clinical and performance evaluation, PMS/PMCF plans, and notified-body interaction records.
- Cybersecurity plus usability evidence scaled to risk.
- UDI records and EUDAMED submission readiness.
Practical notes:
- Small words, big impact: tweaking “intended purpose” can change both your class and the pathway.
- Plan for notified-body expectations on clinical evidence and post-market vigilance early.
- Keep UDI, vigilance, PMS reporting, and PMCF data wired into ops as live systems.
- Track non-EU regulatory divergence (Swissmedic, MHRA) where your roadmap targets markets outside MDR scope.
Decision test: If your “intended purpose” changes by one sentence, do you know exactly how your class and conformity route change?
Emerging technologies in medical device software development
Four emerging technology threads are reshaping medical device software development & test automation: AI/ML for diagnostics and prediction, IoT for continuous monitoring, cloud architectures for scale, and AI-driven test automation for the validation work itself.

AI and machine learning in medical devices: pattern detection at scale
AI and ML sift through large clinical datasets to find patterns and predict outcomes. In medical device software development, the practical applications are diagnostic support, early-warning systems, clinical decision augmentation, and personalized treatment recommendations.
- Predictive analytics: AI algorithms analyze patient data to flag health issues before they become urgent. Heart rate and blood pressure trend analysis can forecast cardiac events, and similar models cover sepsis risk and respiratory decline.
- Personalized treatment plans: Machine learning models tailor treatment plans from individual patient data, increasing the chance the intervention matches the patient.
- Automated diagnostics: AI-driven diagnostic tools analyze medical images and lab results faster than manual review. The result is faster diagnoses with reduced clinician workload on routine cases.
- Controlled model updates: Field data can improve AI performance, but FDA-cleared devices don’t get to “learn in production” freely. Decide early whether a PCCP belongs in the premarket strategy, because that choice shapes the whole change-control path.
- Aidoc’s CARE1, cleared in February 2025, is the first foundation-model-powered clinical AI to receive FDA clearance.
Most cleared AI devices ship with locked models
FDA-authorized AI/ML medical devices are no longer a fringe category. Through the end of 2025, FDA had authorized 1,451 AI/ML-enabled medical devices, with 295 new authorizations in 2025 alone.
Most AI clearances are locked-model 510(k)s with thin clinical evidence
Before 2016, FDA had cleared only 33 AI/ML devices total. The agency now clears roughly that many every six weeks.
The market has less “self-learning AI” than the hype suggests. The dominant FDA pattern is radiology-heavy 510(k) clearance with locked-model deployment. Radiology accounts for about 76% of all FDA AI clearances, and roughly 96% of AI device clearances go through the 510(k) substantial-equivalence pathway.
Clinical evidence for that pipeline is thin. Under 2% of cleared AI devices have RCT backing, and only around 29% of radiology AI clearances included any clinical testing.
AI devices also fail faster after clearance. 43% of AI device recalls occur within 12 months of authorization, roughly twice the rate for all 510(k) clearances.
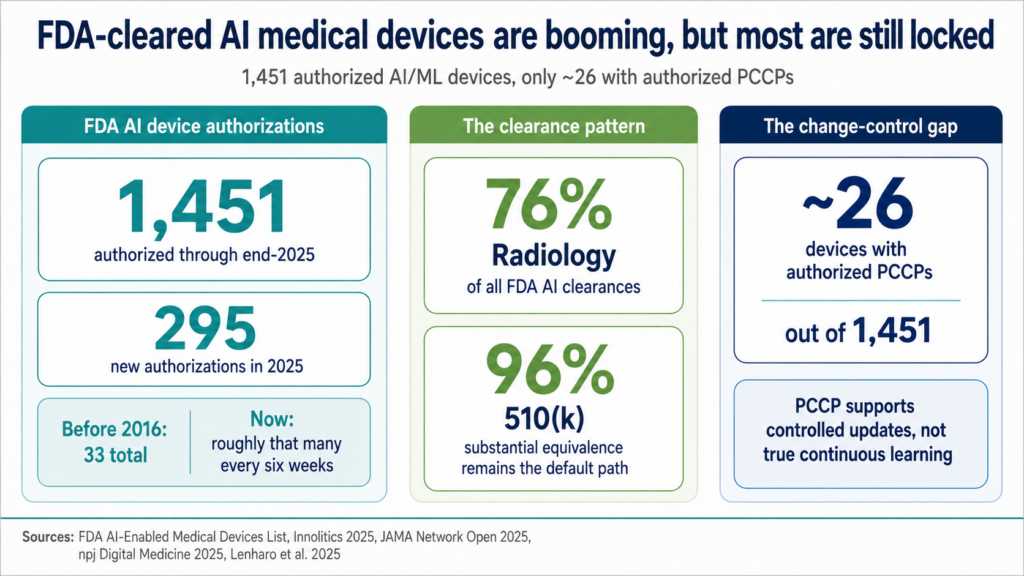
PCCP is FDA’s controlled-change escape hatch, and only 26 cleared devices have one
For AI medical device teams, model improvement only matters if the product has a cleared change-control strategy after launch.
That’s where a Predetermined Change Control Plan, or PCCP, matters. FDA finalized its AI-specific PCCP guidance in December 2024. As of mid-2025, only around 26 of the 1,451 cleared AI devices have an authorized PCCP.
A PCCP lets manufacturers predefine certain post-authorization algorithm changes. The plan spells out the validation protocol and impact assessment needed to make those changes without filing a fresh submission every time. PCCP supports:
- Retraining with new data
- Model changes within the original intended use
- Compatibility updates
- Other changes FDA already accepted inside the PCCP
FDA has still not authorized a truly continuously-learning AI-enabled device. These changes push you back into new-submission territory:
- Changes to intended use
- Expansion to a new patient population
- Increased risk classification
- Anything outside the approved PCCP
For healthcare product teams, PCCP is an architecture decision. Model registries, dataset provenance, rollback plans, bias checks, performance thresholds, release gates, and user communication need to be designed before the first submission. Otherwise, the team ships an AI product that can technically learn, then gets commercially stuck in place.
IoT-enabled medical devices stream patient data continuously
IoT in healthcare means medical devices that collect and transmit patient data continuously. The clinical value is real-time visibility into what’s happening between appointments.
- Remote monitoring: Wearable devices and mobile apps with IoT capabilities monitor vital signs like heart rate and blood pressure, sending data directly to clinicians. This shifts care toward continuous oversight and reduces unnecessary hospital visits.
- Data collection: IoT devices give a continuous, multi-signal view of patient health. The accumulated data feeds software that offers clinical-decision support and treatment recommendations grounded in the patient’s own baseline.

Cloud-based medical software architectures: HIPAA-ready and fleet-deployable
Cloud backends carry what device-side compute can’t: analytics, interoperability, ML workloads, and fleet-wide deployments. Where possible, use:
- HIPAA-ready cloud services (BAAs)
- data-residency controls
- network-segmented environments
- PHI tokenization
Favor event-driven ingestion, FHIR/API gateways for EHR connectivity, containerized services for safe rolling updates, and feature flags that let you cut over without a redeploy. Pair streaming pipelines with model registries so algorithm updates are traceable and roll back cleanly.
Automated testing and validation framework: AI across the development loop
Automation is the highest-impact move in medical device software development & test automation. AI can handle a chunk of the work at each stage: spec drafting, code generation, test case creation, and quality assurance review. The humans focus on judgment calls.
- Automated code generation: AI assists in writing code, reducing manual effort and minimizing human error. The development process moves faster with more consistent output.
- Quality assurance: AI-driven test automation runs comprehensive test cases rapidly, surfacing issues that manual testing tends to miss. Automated testing also keeps the software matched to quality standards and regulatory requirements.
- Spec writing: AI drafts detailed specifications from initial requirements, cutting documentation cycle time and surfacing gaps for human review.
- Traceability automation: AI tools can maintain the requirements → code → test → evidence chain automatically, which is most of what compliance documentation actually looks like.
For budgeting reality on the EHR side of this, see our notes on the cost of EHR implementation.
Key performance indicators for medical device software projects
Medical device software projects need KPIs across two axes: how the build is running, and what it’s doing for patients.

Defining success metrics: the four numbers worth tracking
Concrete success metrics come before the first sprint. Vague goals produce vague projects.
- Time to market: Duration from project initiation to launch.
- Development cost: Total expenditure tracked against budget, with variance analyzed per phase.
- Compliance rate: Adherence to FDA Design Controls and HIPAA, measured by audit findings per release.
- Defect escape rate: Bugs that reach validation or production divided by bugs caught earlier. Lower is better, and trending matters more than the absolute number.
Read more on the medical device cost breakdown
Measuring ROI and patient outcomes: dollars and clinical impact
Value to patients and providers determines whether the build was worth doing.
- ROI: Financial return against development cost, including the post-launch maintenance budget.
- Patient outcomes: Recovery rates, reduced readmissions, improved engagement, and clinician workflow satisfaction.
- User adoption: How widely and frequently the software gets used in the target healthcare setting.
- Time-to-clinical-decision: How fast the software gets a clinician to a confident decision, compared to the workflow it replaced.
Adoption and time-to-decision are the two KPIs that decide whether ROI shows up at all.
For the scheduling side of the workflow, see our notes on medical patient scheduling software.
Leading indicators that predict the lagging ones
All eight KPIs above lag. By the time they move, the project decisions that drove them are already three sprints back. Pair each lagging KPI with a leading indicator the team can act on this week.
Wild velocity swings predict slipped milestones better than the burndown chart. If pilot users can’t finish the priority workflow in usability tests, they won’t in production either. Bundles that don’t generate cleanly today become audit findings later.
Cost considerations for medical device software projects
Costs in medical device software projects show up across four categories: development effort, regulatory compliance, maintenance after launch, and the ROI side that ties spend to outcomes.

The categories below the waterline (compliance, maintenance, security, post-market surveillance) account for most of the lifetime spend. Treat cost as a design constraint from day one.
Development cost tracks risk class and integration scope before headcount
The earlier you lock intended use and IEC 62304 safety class, the clearer your V&V depth and budget become. Class C carries an order of magnitude more evidence burden than Class A. The single largest discrete line item in Class II SaMD development is the summative usability study (IEC 62366-1), which runs $100,000 to $300,000+ for one user group (MD+DI / HirLan, 2022). That’s 4 to 10 times the FDA 510(k) user fee.
Most founders see the $26K user fee and budget accordingly, then discover the usability number midway through the build. eQMS licensing, HITRUST certification, and the year-one cybersecurity build (threat model, SBOM tooling, secure SDLC) round out the meaningful pre-launch line items.
The traps that move budgets: late EHR planning, paper QMS engineers don’t use, bespoke one-offs where a component would do, and underestimating clinical workflow validation time.
Compliance is a recurring line item, budget it like cloud spend
Compliance runs continuously and proportional to risk. It shows up across:
- QMS upkeep: DHF/DMR maintenance, internal audits, CAPA effectiveness checks
- Lifecycle evidence: requirements-to-tests traceability, SOUP inventory, anomaly handling
- Risk file work under ISO 14971
- Usability studies under IEC 62366-1
For EU MDR/IVDR, add notified-body interactions, PMS/PMCF, and clinical/performance eval. For cybersecurity, add coordinated disclosure handling, periodic pen-tests, SBOM maintenance per release, and secure update evidence.
For Class IIb/III devices, notified body annual maintenance alone runs $136,000 to $161,000 per device per year (MedTech Europe IVDR & MDR Survey, 2024). Over a 5-year product lifecycle, that compounds past the FDA PMA user fee.
The cost is built into the regulation: MDR Articles 54, 61, and 83 require continuous clinical evidence and active PMS for every device. None of these end at launch.
Maintenance and update expenses: the long tail that drifts budgets
The long tail is where budgets drift. Platform churn, post-market changes, security hygiene, and dependency rot are all guaranteed.
Post-market actions (safety corrections, field notices, re-validation, PMS reports), platform drift (OS/browser/cloud/EHR version bumps, cert rotation, dependency patching), ML upkeep if AI is in scope (drift monitoring, dataset curation, re-training), and support SLA staffing all show up year after year. If it connects or compiles, it will change. Budget recurring effort accordingly.
Cost ranges, build through sustain
Figures below assume a US-based team at a $128/hr fully-loaded blended rate (BLS OES May 2024 median software developer wage with a 2x multiplier), IEC 62304 Class B as default, and no hardware-in-the-loop. EUR converted at the 2025 IRS annual average ($1.1287 per EUR). Where a row is statutory (FDA fees), the figure is exact for FY2026.
One honest caveat on the table: no clean public IEC 62304 Class A/B/C effort multiplier exists in current literature. Class C carries materially more V&V effort than Class B (Verifysoft, 2024), but the multiplier varies by team and architecture. Rows are scaled to Class B as the default reference point.
ROI calculation methods finance will accept
Finance cares about outcomes. Tie features to defensible cash flows and quantifiable risk reduction. Start with TCO over 3-5 years: development + compliance + cloud/ops + maintenance + support + decommissioning. Then model the benefit categories:
- Revenue (device sales, subscriptions, per-use fees, reimbursable services)
- Cost avoidance (fewer manual hours, reduced rework, lower support tickets, shorter cycle times)
- Risk reduction (fewer incidents/audits, faster approvals via cleaner evidence)
- Speed-to-market (earlier revenue as a function of regulatory pathway and validation overhead)
Express it as ROI = (Cumulative Benefits − TCO) ÷ TCO, with a sensitivity table on the four biggest unknowns: adoption rate, regulatory timeline, integration scope, and dependency risk. Decision test: If a feature can’t be tied to a quantified outcome (revenue, avoided cost, risk reduction, or regulatory headroom) within 12 months, push it out of MVP.
Security considerations in medical device software development
Security in medical equipment software development is where most consequences land for patients, providers, payors, and the company shipping the product.

Cybersecurity risk assessment: model patient-harm threats first
Start with a device-centric threat model (patient harm first), then layer asset inventory, SBOM, coordinated vulnerability disclosure, and incident-response runbook. Tie risks to concrete controls and verification evidence.
The risks that matter for medical devices:
- hacking and unauthorized access to clinical interfaces
- data breach exposure of PHI and patient safety
- supply-chain attacks through SOUP and third-party components
- firmware tampering, replay attacks, MITM on telemetry, and side-channel leaks
Map each one through hazard → threat → control → evidence for every interface (BLE/Wi-Fi/USB/Cloud). Run continuous vulnerability scanning on third-party components and firmware images.
Data encryption and protection: cryptography plus PHI minimization
Protect data in transit and at rest with strong cryptography and sound key management. Minimize PHI by design and segment what remains.
- Encryption: in-transit (mTLS across the device-to-cloud chain, including edge gateway hops) and at-rest, with HSM-backed KMS and key rotation.
- Authentication: MFA for users, SSO/IdP for fleet, short-lived OIDC/OAuth2 tokens, RBAC with least privilege, and JIT elevation for production diagnostics.
- Minimization: tokenize or pseudonymize where full identifiers aren’t needed, and segment what remains.
A compromised credential matters less when access is segmented and tokens are short-lived.
Network security for connected medical devices assumes hostile networks
Assume hostile networks. Isolate clinical traffic, authenticate endpoints, fail safely when connections degrade, and log enough to forensically reconstruct what happened.
- Network segmentation (VLANs/medical DMZ), zero-trust policies, allow-list egress, and inbound traffic inspection at the perimeter.
- Certificate pinning, firmware signing, OTA update integrity checks, and rollback paths if a signed update misbehaves.
- DDoS protection and rate limits at API gateways, plus anomaly detection on telemetry.
- Endpoint authentication on every connection (mutual TLS, no anonymous APIs).
For the integration side of this, see our guide on medical device integration.
Incident response: rehearse the playbook before you need it
Plan for failure like it’s inevitable: detect fast, contain precisely, recover safely, and notify responsibly. Rehearse the playbook.
Build a triage matrix for clinical impact, with clear escalation paths and forensics-ready logging that captures chain-of-custody on the incident evidence. Immutable backups and staged rollbacks for firmware and app give you safe recovery paths. Post-incident CAPA closes the loop, and a notification path reaches affected clinical sites.
Third-party security integration: vendors expand your attack surface
Vendors and dependencies expand your attack surface. Treat them as part of your system from the start.
- BAA-backed services for any PHI touch; verify their audit reports and patch cadence.
- SSO/IdP hardening (SAML/OIDC), MDM for fleet control, secure EHR integrations, and quarterly access reviews.
- Third-party SBOMs, contractually required vulnerability disclosures, sandboxed data exchanges, and incident-response coordination clauses in contracts.
Topflight’s compliance strategies bake audit-readiness into every sprint
Security compliance and regulatory compliance feed one audit trail. The practices below build it during the SDLC, so the bundle is ready when submission week comes.
Risk analysis runs with the client before sprint one, with each design decision traced to the hazard it mitigates. Reconstructing that mapping after a build is shipped is harder than it looks, and the result rarely matches the codebase.
We document the rationale for every formal step deferred. If a project skips formative HFE for now, the file explains why SME-led usability interviews were sufficient at this stage. That document is what the auditor needs when the framework shifts again, and it shifts.
Regulated scope stays isolated from non-clinical features:
- wellness layers
- admin portals
- content management systems
- analytics dashboards
The non-clinical parts ship at startup speed while the device-side carries full 62304/14971/62366/13485 discipline.
Continuous monitoring with real-time security alerts wires to on-call. We tune thresholds monthly to catch baseline drift before it surfaces as an alert storm. Vendor and third-party audit results get reviewed quarterly, with revocation paths gated on SOC 2 / HITRUST currency and a working coordinated-disclosure channel.
By submission week, the audit trail is already done.
Future trends in medical device software development
Five healthcare technology threads will reshape medical device software development over the next 5 years: AI for diagnostics, blockchain for tamper-evident records, 5G for high-fidelity remote telemetry, quantum computing for optimization and cryptography, and digital twins for scenario testing.

AI-powered diagnostic software raises the ceiling on clinical accuracy
AI will keep raising the ceiling on clinical accuracy and speed, especially where pattern recognition and triage matter most.
- Better diagnostic tools driven by image analysis and pattern detection.
- Predictive analytics for patient care and early-warning systems.
- Automation of routine clinical tasks like lab result review, charting, triage queuing, and prior-auth packet prep.
- Algorithmic decision support tied to evidence and traceable model versions.
The version that gets deployed safely is the one with a working PCCP and rollback path.
Blockchain for medical device data: tamper-evident logs and chain-of-custody
Use blockchain selectively in medical device scenarios where multiple parties need to verify the same data without trusting each other. Tamper-evident logs, chain-of-custody, multi-party data exchange, and cross-vendor audit trails are the classic use cases.
Treat it as a complement to existing standards. The applications that justify it:
- tamper-evident audit trails across vendors
- cryptographic verification on real-time data sharing across platforms
- standardized data formats and provenance markers
- cross-organization audit trails without a central trusted party
For everything else, traditional databases with signing and logging do the job faster and cheaper.

5G-enabled remote healthcare devices bring intervention to the edge
5G enables higher-fidelity telemetry and near-real-time interventions at the edge. Useful for imaging, wearables, at-home diagnostics, and continuous wearable monitoring during clinical trials.
- Remote monitoring through mobile apps streamed over 5G, with single-digit-millisecond round-trip latency on critical alerts.
- Bandwidth headroom for continuous imaging telemetry from at-home equipment.
- Edge inference for time-sensitive clinical analytics, with results processed locally for faster turnaround.
The latency drop turns chronic monitoring from observation into intervention.
Quantum computing in medical software: exploratory now, crypto-agility soon
Near-term impact stays exploratory across combinatorial problem-solving, molecular simulation, cryptography resilience, and drug discovery support. Track vendor roadmaps while designing crypto-agile systems so upgrades don’t require rewrites.
Digital twin technology for medical devices enables scenario testing before patient contact
Patient- and device-level digital twins enable scenario testing, therapy tuning, maintenance scheduling from telemetry, and field-readiness simulations before any change touches a real patient or device.
- Therapy parameters tuned per patient against twin-simulated response curves before any dose change.
- Predictive maintenance scheduling driven by device-level twin telemetry, ahead of physical wear surfacing as failure.
- Field-readiness simulations for firmware and hardware changes before staged rollout.
- Faster regulatory submission packages via in-silico validation, with reproducible evidence the auditor can replay.
For the regulatory path on AI and emerging-tech features, see our notes on SaMD certification.
How Topflight helps you build medical device software
Our medical device software development work spans computer-vision remote patient monitoring and IoT-connected consumer hardware. Each software product below shipped through its own regulatory path. Three recent projects:
AllHeartz
AllHeartz turned the smartphone into a clinical-grade sensor using computer vision. It’s a remote therapeutic monitoring (RTM) app that gives clinicians accurate movement data without an in-person visit.
- Used AI to cut in-person visits by up to 50% and clerical work by 80%.
- Enabled patients to run at-home exams through the mobile app, capturing movement data at clinical-grade accuracy.
- Implemented real-time pose recognition with interactive charts feeding the clinician dashboard.
- Built a cloud-based computer vision platform that scales with patient volume.
- Achieved HIPAA compliance with data encryption and secure connections.
Explore the AllHeartz Case Study
Dedica Health
For Dedica Health, we built a web platform integrated with clinically certified medical sensors. To make it work, we had to develop medical device software that hooks into existing medical systems and supports continuous data collection and real-time monitoring.
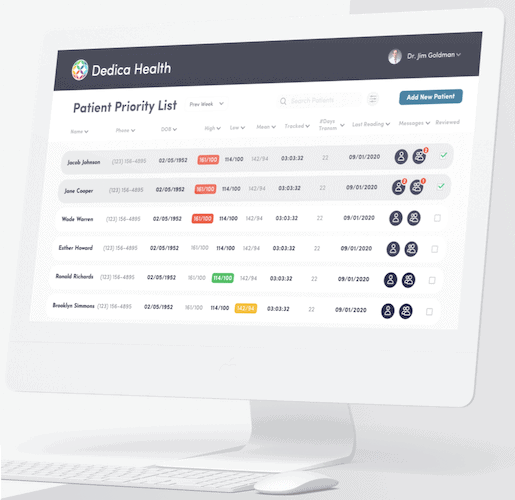
- Monitored over 1,100 patients daily, generating a $300,000 ARR deal through a SaaS model.
- Simplified operations with automated routines and health data visualization.
- Integrated clinically certified sensors for accurate data collection and patient monitoring.
- Used rapid prototyping and continuous development to iterate fast on features.
- Built a secure platform with documented safeguards covering encryption, access control, audit logging, and breach-detection tooling.
Explore the Dedica Health Case Study
Joovv Light Therapy App
For Joovv, we built a mobile app that connects to their hardware fleet across legacy and current device models. The app gives users control and monitoring through a smartphone interface, and gives Joovv a marketing-analytics layer on top.
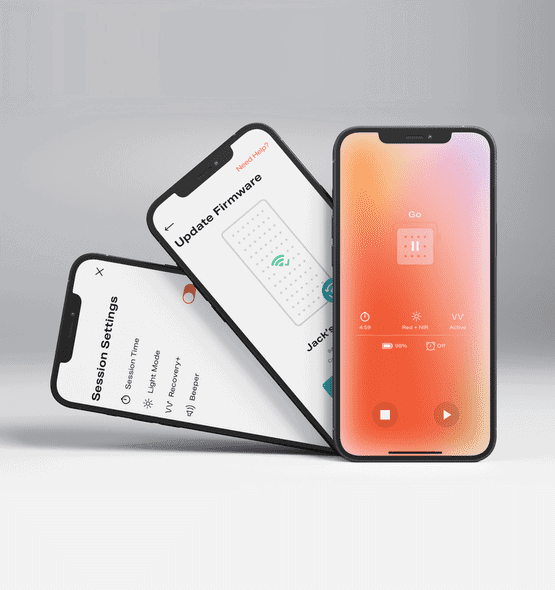
- Rebuilt the app to support new hardware, with user-engagement features around goal setting and session recovery.
- Ensured compatibility with both legacy and new devices using React Native.
- Developed a web portal for user management and app usage metrics that feed marketing efforts.
- Achieved HIPAA compliance and adhered to IEC 62304 standards for IoT medical software.
Explore the Joovv Light Therapy App Case Study
We’ve shipped medical device software through FDA submissions and HIPAA audits. As a software development company built around healthcare workflows, we plug in as a development partner from concept through post-market. Our software development services cover the lifecycle end to end.
Schedule a call to talk through where your build sits today.
For general-purpose hardware running a clinical workflow, we ship software solutions that hold up in audit.
Frequently Asked Questions
What are the phases of medical device development?
Five phases: concept and feasibility, design and development, verification and validation, regulatory approval, then deployment and maintenance. The two most underestimated are verification (where edge cases surface late) and post-market maintenance (where most of the long-term software cost lives).
What tech stack do you use for medical device software development?
It depends on where the software runs. Embedded firmware is usually C or C++. Mobile companion apps run Swift on iOS and Kotlin on Android. Backends use Python or Node on AWS or Azure, with SQL databases. Web dashboards lean on React or Angular.
What is the cost of building a medical device software?
Costs swing on complexity, regulatory requirements, the development team, and how much hardware integration is in scope. Plan on $100,000 to several million dollars for a market-ready product. The good news: you can validate the core software and prove traction on a much leaner budget before committing the full regulatory spend.
What are some examples of medical devices?
Pacemakers, insulin pumps, MRI machines, and wearable health monitors. Each ships with embedded firmware that drives device function plus a companion layer (mobile app or clinician dashboard) that ties into patient care workflows. The companion layer is usually where the software work concentrates.
How long does it usually take to create medical devices?
Timelines run from 18 months to several years. The drivers: device classification, FDA submission pathway, clinical trial scope, and any hardware that needs verification testing.