
If you prescribe peptides right now, the patient conversation is moving faster than the legal infrastructure.
The July 23 to 24, 2026 PCAC meeting could change the compounding pathway for seven peptides: BPC-157, KPV, TB-500, MOTS-c, Emideltide/DSIP, Semax, Epitalon. It won’t change what you owe the patient. This peptide therapy physician guide covers both sides of that line.
If FDA follows a positive PCAC recommendation and completes rulemaking, 503A pharmacies will have a legal pathway to compound these substances for individual patients. Your obligations on informed consent, pharmacy verification, documentation, and malpractice exposure remain identical regardless of outcome.
What is settled: the meeting will happen, the seven peptides are on the agenda, and the docket is open for public comment. What remains unsettled: whether FDA will follow PCAC’s recommendation, and how long rulemaking will take afterward.
If you’re already prescribing, or considering it, the question is the same: how do you operate compliantly while the legal pathway is still being built?
What does the July 2026 PCAC meeting change for physicians evaluating peptide therapy?
A positive recommendation followed by FDA rulemaking would create a legal 503A compounding pathway for the seven peptides under review (BPC-157, KPV, TB-500, MOTS-c, Emideltide/DSIP, Semax, Epitalon). The compounding pathway sits separately from prescriber obligations on informed consent, pharmacy verification, documentation, and malpractice exposure. Those obligations apply to any unapproved bulk drug substance.
Key Takeaways:
- The PCAC outcome affects the compounding pathway alone. A positive recommendation followed by FDA rulemaking would create a 503A path for the seven peptides under review; consent, pharmacy verification, documentation, and malpractice exposure remain identical regardless.
- Most current peptide prescribing sits outside the legal compounding framework, and enforcement is tightening. The March 2026 Peptide Sciences shutdown, the 40-state-AG letter, and new telehealth rules close the gray zone fast.
- Practice infrastructure has to be built before the first prescription. Structured intake, verified pharmacy integration, async communication, and cash-pay billing are the four requirements. Off-the-shelf platforms handle the easy 10%; the hard 90% has to be built.
Table of Contents
- What the PCAC meeting could change and what it cannot
- Most peptide prescribing today happens outside the legal compounding framework
- The evidence base, what physicians need to know before prescribing
- Informed consent for compounded peptide prescribing
- Compounding pharmacy verification, what physicians must confirm
- Practice infrastructure, four requirements before the first patient
- Medical board and malpractice exposure, an honest assessment
- How Topflight Apps can help
What the PCAC meeting could change and what it cannot
A positive PCAC recommendation followed by FDA rulemaking would create a legal 503A compounding pathway for the listed peptides. It wouldn’t grant FDA approval, establish efficacy, eliminate prescription requirements, or change what the prescribing physician owes the patient.
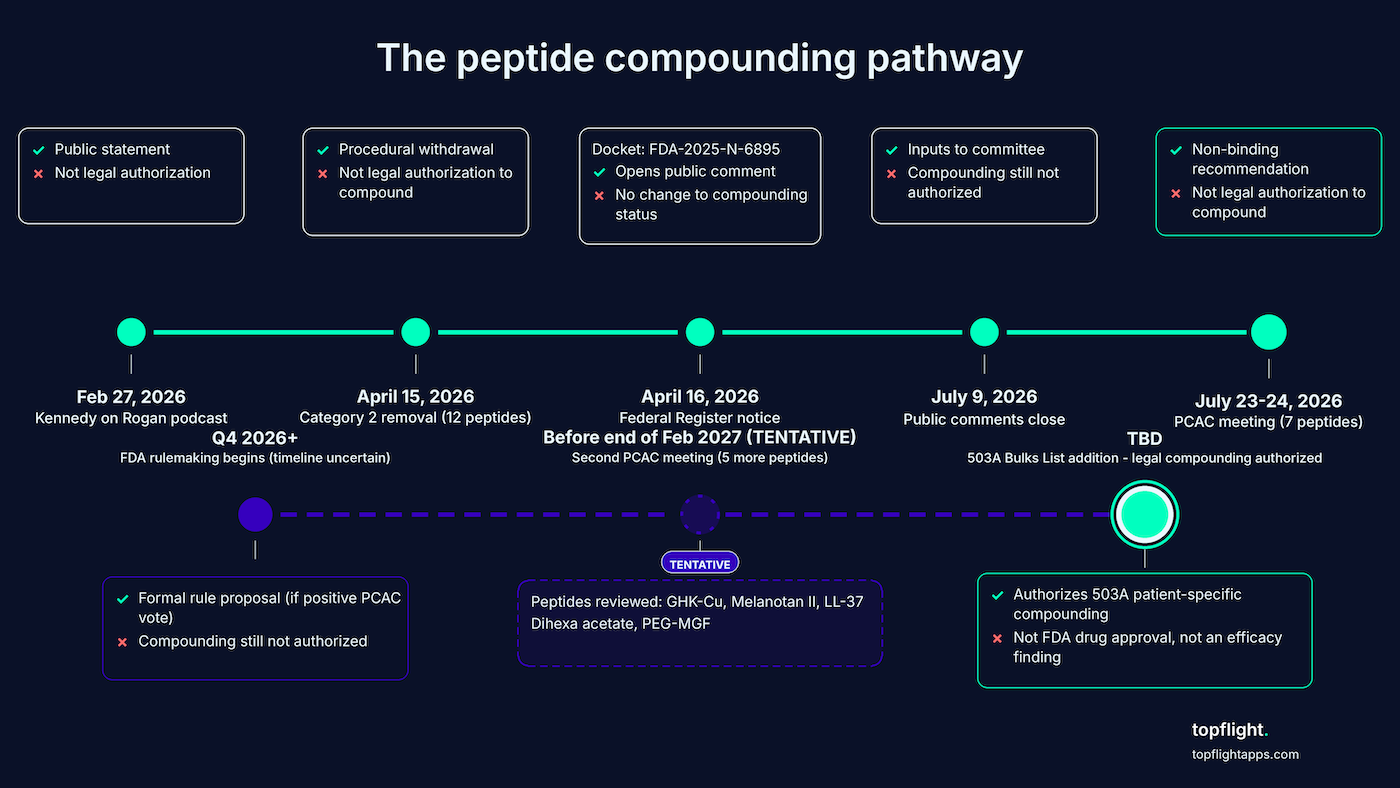
What “inclusion on the 503A Bulks List” actually authorizes
Inclusion on the 503A Bulks List means FDA has determined, through formal rulemaking, that a bulk drug substance may be used in 503A patient-specific compounding. It’s not FDA drug approval. The FDA peptide compounding decision 2026 turns on this single regulatory step, and it’s a process most physicians have never had to track.
The seven peptides on the Section 503A Bulk Drug Substances List agenda split across the two meeting days:
- Day 1 (July 23): BPC-157 (ulcerative colitis), KPV (wound healing and inflammatory conditions), TB-500 (wound healing), MOTS-c (obesity and osteoporosis).
- Day 2 (July 24): Emideltide / DSIP (opioid withdrawal, chronic insomnia, narcolepsy), Semax (cerebral ischemia, migraine, trigeminal neuralgia), Epitalon (insomnia).
The Federal Register notice (Docket No. FDA-2025-N-6895) opened the public comment window. FDA still considers comments filed between July 9 and July 22. Oral presentations require notification by June 30, 2026.
The procedural removal in April was not the green light
The substantive review happens at the PCAC meeting in July. April was a procedural step.
On April 15, 2026, FDA removed these seven peptides from 503A Category 2 by withdrawing their nominations. National Law Review (Polsinelli PC) was explicit on April 22, 2026: removing a Category 2 bulk drug substance “does not, on its own, authorize use of that substance in compounding or bring it within FDA’s interim enforcement discretion policy for substances in Category 1.” Even Category 1 substances are only under enforcement discretion, which is short of full authorization.
PCAC recommendations are non-binding. FDA must then initiate formal rulemaking to add a substance to the 503A Bulks List, which takes additional months after a positive recommendation. A second PCAC meeting before the end of February 2027 will review five additional peptides:
- GHK-Cu (injectable routes only; non-injectable routes are moving back to Category 1 directly)
- Melanotan II
- Cathelicidin (LL-37)
- Dihexa acetate
- Mechano Growth Factor Pegylated (PEG-MGF)
What does not change regardless of outcome
The prescriber’s obligations don’t move with the regulatory status of the compound.
- Prescription requirement: these remain prescription-only compounded drugs.
- Physician-patient relationship requirement: a valid clinical relationship must be established before prescribing.
- State prescribing authority and telehealth standards: vary by state, must be followed.
- Informed consent, documentation, and liability exposure for adverse outcomes all carry forward unchanged. We cover each in Sections 4, 6, and 7.
- Malpractice coverage terms don’t change automatically. Verify with your carrier.
What remains unsettled
Several pieces of the picture aren’t yet fixed.
- Individual peptides may receive different PCAC recommendations (Day 1 and Day 2 are reviewed separately).
- FDA may not follow PCAC’s recommendation under the Drug Quality and Security Act framework.
- Rulemaking timelines are unpredictable.
The committee itself is significantly understaffed. As of May 2026, PCAC’s public roster shows only three voting members of the twelve required: Timothy D. Fensky, RPh; Elizabeth Rebello, RPh, MD; and Brian Serumaga, PhD.
The chairperson and consumer representative seats are vacant. Kennedy may appoint new members before July, and committee composition will shape the recommendation. So be explicit with patients and with yourself: physicians prescribing now based on the April Category 2 removal are operating without a clear legal compounding framework.
Most peptide prescribing today happens outside the legal compounding framework
What was announced in February 2026 isn’t what’s legally authorized today, and the gap is the reason compliance-first physicians are right to wait.
The Kennedy announcement wasn’t the green light it sounded like
On Joe Rogan’s podcast in February 2026, HHS Secretary Robert F. Kennedy Jr. said he expected FDA to make “about 14” Category 2 peptides “more accessible” within “a couple of weeks.” The April action delivered 12. What was marketed as “BPC-157 is back” isn’t legally true. Kennedy himself called April 15 an action that “begins to restore regulated access.” The emphasis sits on “begins.”
Where prescribing compounded peptides actually happens today
Most physicians prescribing compounded peptides today work outside the legal framework: research-chemical vendors with “research use only” labeling, telehealth peptide clinics citing the April Category 2 removal as authorization (regulatory commentators have flagged this as not authorization), medical tourism, and wellness clinics marketing “hormone optimization” programs that imply peptide use without naming compounds.
FDA’s March 14, 2025 Final Guidance on Research-Grade Biological Compounds presumes residential shipping or dosing instructions equal intent for human use regardless of labeling. The kind of IoT telehealth integration that closes the loop between intake and outcome monitoring is what separates a compliant practice from the consumer-facing stacks these channels run on.
The enforcement climate is tightening
Peptide Sciences, the largest gray-market research peptide vendor in the U.S., voluntarily shut down on March 6, 2026. In 2025, more than 40 state attorneys general formally letter FDA about products with unknown contaminants and online sales of “research use only” GLP-1 active ingredients sold without prescriptions.
State AGs use consumer protection law to pursue sellers FDA may not prioritize, and “research use only” labels don’t provide legal protection when the product is clearly marketed for human consumption. Useful framing from Dr. Christopher Mendias in Medscape (May 1, 2026): treat the peptide conversation “like an illicit drug use conversation, eventually moving to a gentle ‘follow-the-money’ discussion.”
The evidence base, what physicians need to know before prescribing
Before prescribing any of these compounds, you have to articulate clinical reasoning, document the evidence you relied upon including its limitations, and demonstrate informed consent occurred. The evidence base is thinner than the popular discourse suggests.

Day 1 peptides (BPC-157, KPV, TB-500, MOTS-c)
The four Day 1 candidates share one feature: their clinical evidence base is significantly thinner than their consumer profile.
BPC-157
Synthetic 15-amino acid peptide derived from a protein in human gastric juice. Studied primarily in animal models, with limited human clinical data. Not approved by any drug regulatory agency for any human use, and on the WADA prohibited list. FDA’s 2023 Category 2 placement cited immunogenicity risk for certain routes of administration, peptide-related impurities, complexities in active pharmaceutical ingredient (API) characterization, and limited safety information for the proposed routes. FDA’s October 2024 PCAC briefing document is the most authoritative summary of available evidence. Start there. Under PCAC review for ulcerative colitis.
KPV
Tripeptide. C-terminal fragment of alpha-MSH. Earlier-stage evidence base than BPC-157. FDA’s 2023 Category 2 placement cited absence of human exposure data. Under PCAC review for wound healing and inflammatory conditions.
TB-500
Synthetic heptapeptide corresponding to the active fragment of thymosin beta-4. Primarily encountered as a designer drug in racehorses. No FDA-approved human use. WADA prohibited. No rigorous human clinical trials. Under PCAC review for wound healing.
MOTS-c
Mitochondrial-derived peptide with obesity and metabolic research interest. Earlier-stage evidence base. FDA’s 2023 Category 2 placement cited significant immunogenicity risk for some routes of administration, plus impurity and API characterization concerns. Limited human data. Under PCAC review for obesity and osteoporosis.
Day 2 peptides (Emideltide, Semax, Epitalon)
The Day 2 peptides have even less Western clinical infrastructure behind them than the Day 1 group.
Emideltide (DSIP, delta sleep-inducing peptide)
Studied for sleep and opioid withdrawal applications. Limited contemporary human data. Under PCAC review for opioid withdrawal, chronic insomnia, and narcolepsy.
Semax
Developed in Soviet-era Russian neuropeptide research. Registered in Russia with ATC code N06BX. Not FDA-approved, and not commercially available in the U.S. through standard channels. Some human clinical data from Eastern European literature for neurological indications, not replicated in Western regulatory-standard trials. Under PCAC review for cerebral ischemia, migraine, and trigeminal neuralgia.
Epitalon
Tetrapeptide derived from pineal gland research. Small human clinical studies in the published literature primarily from Russian researchers (retinitis pigmentosa, pulmonary tuberculosis). No Western regulatory-standard trials, and no FDA approval. Under PCAC review for insomnia (per Federal Register notice, FR Doc. 2026-07361).
The prescriber’s obligation when evidence is thin
The standard of care for any compound with a limited human evidence base is consistent across these seven: thinner evidence equals heavier documentation. You have two non-negotiables. A documented clinical rationale per patient. A documented informed consent discussion (next section). Patient selection criteria belong in the chart.
BPC-157 physician prescribing carries the most consumer awareness and arguably the most scrutiny, given how openly it circulates on Reddit and X. The discipline applies equally to all seven unapproved bulk drug substances regardless of cultural visibility. The chronic disease management app infrastructure most practices already use was built around FDA-approved drugs with established billing codes, which means the documentation layer for unapproved peptides has to do work the EHR alone won’t do.
Informed consent for compounded peptide prescribing
No appellate court has ruled that off-label disclosure is required as part of informed consent, but that precedent only addresses off-label use of FDA-approved drugs. For unapproved compounds like these peptides, the judicial picture is even less settled. Documented consent is your primary defense in a board investigation or malpractice claim involving any unapproved compound.
Standard off-label framing doesn’t apply here
Off-label prescribing of FDA-approved drugs is legal and common, with case law on what disclosure is required when an approved drug gets used for a non-approved indication.
These peptides are unapproved bulk drug substances with no approved indication for any of them. The off-label framework doesn’t transfer because there’s no approved-drug anchor to start from. The consent has to characterize the compound accurately as an unapproved bulk drug substance prescribed through a compounding pharmacy. Don’t frame it as “approved for X, using for Y.”
The legal framing here comes from published medical liability analysis and psychiatry-law literature on informed consent and the physician-patient relationship.
What the consent discussion must cover
The consent discussion has to cover six specific elements before the prescription is written.
- The compound isn’t FDA-approved for any human use, in any indication.
- The evidence base for the proposed use and its limitations: primarily preclinical, or limited human studies not conducted under Western regulatory standards.
- Known and theoretical risks, including those that may not be fully characterized given the limited human data.
- Alternatives the physician considered, including the option of no treatment.
- That the patient may decline or discontinue at any time.
- The monitoring plan: what you’ll track, and what adverse events should prompt the patient to contact the practice.
Documentation requirements
The compounded peptide informed consent has to be written, signed, in the medical record, and refreshed as the regulatory picture moves.
- Written and signed in the medical record. Verbal discussions and checkbox-only forms don’t survive a board investigation. Many practices capture consent digitally now, where the HIPAA-compliant app development layer that handles intake also handles consent capture with audit-grade timestamps.
- Per established liability guidance, the worst outcome in a board investigation is a patient who says the physician “never even told me this wasn’t approved.”
- For long-term prescribing, re-obtain and re-document consent as the evidence base and regulatory status evolve, particularly if FDA acts on the PCAC recommendation and the compound’s legal status changes.
Document the clinical rationale per patient in the chart itself: why this compound, why this patient, what you expect to monitor. The chart is the audit trail.
Compounding pharmacy verification, what physicians must confirm
Compounded drugs aren’t subject to the GMP requirements applicable to commercial manufacturers, and variability in strength, sterility, and stability has been documented. Verification is your responsibility; pharmacy marketing copy isn’t a substitute. The prescriber-pharmacy integration layer that automates this verification at scale is usually built by external healthcare app developers rather than handled inside the EHR itself.
503A vs. 503B, what the distinction means for prescribers
A 503A compounding pharmacy compounds for individual patient prescriptions under state board of pharmacy oversight. If a peptide reaches the 503A Bulks List after PCAC recommendation and FDA rulemaking, 503A pharmacies may compound it under patient-specific prescription.
A 503B outsourcing facility registers with FDA, operates under cGMP, and produces larger batches for distribution to healthcare providers without patient-specific prescriptions. Quality standards run higher than 503A’s. The tradeoff is fewer facilities and more limited formularies.
For sterile injectables, 503B outsourcing facilities offer higher quality assurance. Confirm whether your pharmacy is 503A or 503B.
What to request and verify
Six items go on the verification checklist before you write the first prescription.
State pharmacy board licensure and standing
Verify good standing in the pharmacy’s home state and any state where it ships to your patient.
Certificate of analysis (CoA) for the specific batch
Documents potency, purity, and sterility testing results. Request and review before dispensing. A CoA reflects what was tested. What the patient actually receives depends on storage and handling after that.
Bulk API sourcing
Confirm the pharmacy’s API sourcing and whether suppliers are on FDA’s approved supplier list. FDA has flagged bulk substances marketed as “research grade” or “research use only” as heightened regulatory and quality risk.
USP 795 (non-sterile) and USP 797 (sterile) compliance
Ask the pharmacy directly.
PCAB (Pharmacy Compounding Accreditation Board) accreditation
Third-party signal of quality standards beyond minimum state licensure.
Sterility testing methodology and beyond-use dating
Red flags include pharmacies that can’t or won’t provide CoAs, pharmacies in jurisdictions without compounding pharmacy licensure requirements, and pharmacies selling directly to consumers without prescriptions.
State variation
State law varies and moves fast.
California’s October 1, 2025 Board of Pharmacy rules permit sterile compounding pharmacies (resident and non-resident) to compound with 503A Bulks List substances under USP 797, reversing the prior prohibition. Pharmacists carry an affirmative duty to verify and document patient-specific “clinically significant differences” from approved alternatives.
Florida’s SB 860 and HB 877, filed in December 2025, would impose API-sourcing and documentation conditions on distribution of certain compounded drugs. Both remain in committee as of early 2026.
Ohio’s February 2026 peptide compounding guidance added state-specific documentation mandates. Recent enforcement includes a Columbus pharmacy that agreed to a $15,000 fine for preparing bulk semaglutide without patient-specific prescriptions.
Louisiana SB 253 (effective August 1, 2026) is the most permissive framework so far. Once a peptide is removed from FDA Category 2, state-licensed pharmacies may compound it provided the API comes from an FDA-registered manufacturer and other federal and state requirements are met.
Consult your state pharmacy board in addition to this guide.
Practice infrastructure, four requirements before the first patient
Four infrastructure elements define peptide therapy practice readiness, and all of them have to be in place before the first patient. Structured intake with audit trail. Verified compounding pharmacy relationship. Async patient communication and outcome monitoring. Cash-pay billing that doesn’t create insurance fraud exposure.
1. Structured intake and audit trail
Structured intake means every patient touch creates a discoverable, timestamped record in the medical chart.
- Minimum clinical documentation per patient: history and physical relevant to the indication; review of contraindications and drug interactions to the extent known for these compounds; documentation that informed consent occurred and what it covered; clinical rationale for the specific compound and formulation.
- Telehealth prescribing standards. The Ryan Haight Act applies to controlled substances; these peptides aren’t currently scheduled. State telehealth prescribing standards apply and vary. Confirm your state’s requirements for establishing a valid prescriber-patient relationship via telehealth before writing any prescription.
- Tightening remote-prescribing oversight. New York finalized rules in May 2025 governing prescription of controlled substances through telemedicine. Peptides aren’t controlled, but the rule signals direction of travel for remote prescribing.

The audit trail has to be producible in a board investigation. Electronic health records with timestamped entries are the standard, and the EHR layer is the natural place where HIPAA-compliant software development principles meet day-to-day clinical documentation.
2. Verified compounding pharmacy relationship
Establish the pharmacy relationship up front, before you write the first prescription. Confirm 503A vs. 503B, CoA availability, API sourcing, and state licensure (Section 5 carries the verification checklist; no need to repeat it here).
E-prescribe compound Rx capability matters next. Standard e-prescribing platforms on the Surescripts network handle most compound Rx; confirm the platform supports the specific formulations you intend to prescribe. Compounding pharmacy integration runs multi-channel at the better partners. Restorative Compounding, for example, licensed in 38 states, offers:
- API integration
- eRx
- eFax
- provider portal submission
Match the integration to your EHR’s capabilities. For refill and monitoring workflow, structure prescriptions for a defined monitoring interval. Avoid open-ended refills for compounds with limited evidence bases.
3. Patient communication and outcome monitoring
Outcome tracking is both ethical obligation and liability protection given the limited human evidence base.
Outcome tracking
Document baseline assessments, the specific outcomes you’re monitoring, and follow-up findings at each interval. Remote patient monitoring app development principles apply for any device-feed integration that pulls patient-reported or biometric data back into the chart.
Adverse event monitoring
Document each adverse event the patient reports together with your clinical response and any FDA MedWatch reporting (voluntary for physicians, required for manufacturers).
Async patient communication via a HIPAA-compliant channel
Any digital channel that touches patient PHI requires HIPAA-compliant communication. Standard consumer messaging apps (SMS, WhatsApp) don’t clear that bar without a BAA.
Clinical documentation in the medical record carries the audit trail
Per-encounter notes link back to consent, clinical rationale, adverse-event context, and outcomes over time.
4. Cash-pay billing
Insurance doesn’t cover compounded peptides because no FDA-approved indication and no billing code exist for them. Billing a health insurer for a non-covered compounded drug is a fraud and abuse risk. Plan accordingly: this is a cash-pay practice model.
Compounded peptides typically cost $100 to $400 per month patient out-of-pocket depending on compound and dosing. Document the fee arrangement in writing before treatment begins, and give the patient a written fee schedule.
For subscription billing models on ongoing prescribing, consult a healthcare attorney before implementing. State fee-splitting and kickback rules vary, and the subscription structure that works in one state may trip another.
Medical board and malpractice exposure, an honest assessment
In a board investigation or malpractice claim, the file you can produce matters more than the evidence base behind the compound.
The pattern that attracts board scrutiny
State medical boards audit for thin documentation. The presence of unapproved compounds in a chart isn’t itself the trigger.
State medical board audits of peptide-prescribing practices have increased in several states. The pattern that attracts medical board scrutiny: prescribing without a documented clinical rationale or informed consent file, often with no follow-up monitoring either.
The New York Office of Professional Medical Conduct (OPMC) actively investigates physicians prescribing unapproved substances. Documentation and informed consent face high-standard review, and the clinical rationale behind each prescription has to hold up under that review.
The Florida Board of Medicine and Florida Department of Health require legitimate medical purpose, proper documentation, patient evaluation, and off-label prescribing that meets accepted standards of care. Failure on any of these can trigger disciplinary action even if federal laws aren’t directly violated.
The pattern is consistent across boards: thin documentation invites scrutiny regardless of clinical outcome.
Your malpractice carrier may not cover this
Malpractice coverage for compounded drug prescribing varies by carrier. The compounded medication risk profile is far enough from standard pharmaceutical malpractice that some carriers exclude it entirely.
The Cooperative of American Physicians, in February 2026, was explicit: “Confirm that your professional liability coverage applies to the prescribing or manufacturing of compounded medications” before doing so.
The worst-case pattern is a patient harmed by an unapproved peptide and an insurer who declines coverage because the treatment fell outside the standard of care or federal compliance. Get the answer in writing from your carrier before prescribing.
The Department of Justice has prosecuted compounders for distributing unapproved peptides, resulting in fines and asset forfeiture. Physician exposure is separate but adjacent to compounder exposure.
Documentation is the only real defense
Documentation defends both the board investigation and the malpractice claim.
Per published NIH/PMC guidance: “Thoughtful documentation of good clinical care and the informed consent process will discourage many plaintiffs’ attorneys from accepting a case. Conversely, poor or no documentation of even the best clinical care can make you an attractive target.”
Documented clinical rationale plus documented informed consent equals the primary defense in both board investigations and malpractice claims. Compounded peptide prescribing compliance turns on this in practice, and the same documentation work limits prescriber liability for the long tail of complications that may surface months after the prescription. Thinner evidence requires heavier documentation: prescribing a compound with minimal human evidence for an indication not supported by that evidence carries different legal weight than prescribing an established compound with a literature base.
The practical takeaway is unromantic. Documentation is the workflow.
How Topflight Apps can help
This piece has been for physicians evaluating their compliance obligations. The infrastructure conversation is for physician-operators, practice owners, and technical leads building compliant peptide practices.
Honest tradeoff: most off-the-shelf telehealth platforms handle the easy 10% (video, scheduling, basic intake) and leave the hard 90% (compound Rx workflow, audit-grade documentation, multi-state licensure tracking, outcome capture for limited-evidence compounds) for the practice to figure out. We’ve watched this pattern across dozens of healthcare builds.
What we build for this use case:
- Structured clinical intake with audit-grade documentation
- Compounding pharmacy e-prescribe integration (multi-channel: API, eRx, eFax, portal)
- HIPAA-compliant async patient communication and outcome tracking
- Cash-pay subscription billing architecture that doesn’t create state-law exposure
If you’re evaluating the technology infrastructure for a compliant peptide therapy prescribing practice, talk to us about your workflow requirements before committing to a platform. The platform decision is downstream of the workflow decision, and the workflow decision is downstream of the compliance posture you’ve chosen. Get the order right and the build comes in cheaper and easier to defend in audit.
FAQ
Is prescribing BPC-157, TB-500, or the other peptides under review legal right now?
No clear legal compounding pathway exists. The April 15, 2026 removal from Category 2 was procedural; the substantive review happens at the July 23 to 24, 2026 PCAC meeting, and FDA must then act through formal rulemaking. Physicians prescribing these compounds now operate without a clear compounding framework.
What informed consent documentation do I need before prescribing a compounded peptide?
Written, signed consent in the medical record. The consent must accurately characterize the compound as an unapproved bulk drug substance (not as an approved drug used off-label), the evidence base and its limitations, known and theoretical risks, alternatives considered, the monitoring plan, and the patient’s ability to decline or discontinue. Re-consent as regulatory status evolves.
Can I prescribe compounded peptides via telehealth?
State telehealth prescribing standards apply. The Ryan Haight Act covers controlled substances, and these peptides aren’t currently scheduled. Every state has its own requirements for establishing a valid prescriber-patient relationship via telehealth, and oversight is tightening (New York’s May 2025 telehealth controlled-substance rules signal the direction). Confirm your state requirements before prescribing.
Will my malpractice insurance cover compounded peptide prescribing?
Coverage varies by carrier. The Cooperative of American Physicians advises confirming with your carrier before prescribing compounded medications. Standard policies may exclude treatments that fall outside the standard of care or federal compliance, which could apply to peptides prescribed before a 503A pathway is established. Get the answer in writing.
What happens after PCAC makes a recommendation, when would compounding actually become legal?
PCAC recommendations are non-binding. FDA must then initiate formal rulemaking to add a substance to the 503A Bulks List, which takes additional months. A positive recommendation on July 24, 2026 does not translate to legal compounding on July 25. Plan for a gap between the recommendation and regulatory effect.
What billing code do I use for a compounded peptide prescription?
There isn’t one. These compounds have no FDA-approved indication and no billing code, and insurance does not cover them. Bill the patient directly as cash-pay; billing a health insurer for a non-covered compounded drug creates fraud and abuse exposure.