
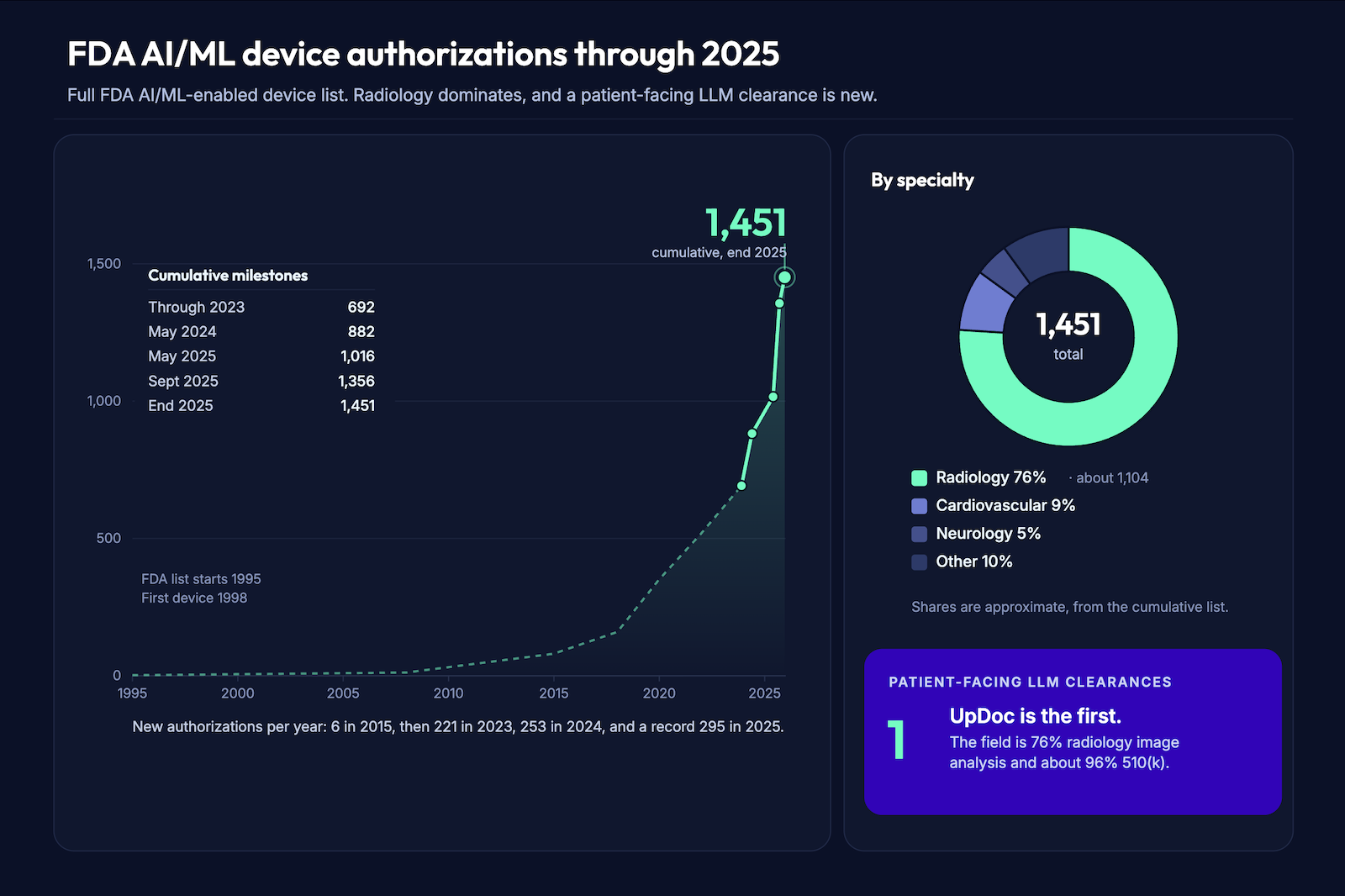
On June 25, 2026, UpDoc announced an FDA 510(k) clearance (K253281) it had received on December 23, 2025, for what it calls the first software as a medical device built on patient-facing large language models. The clearance covers insulin and medication management for adults with type 2 diabetes, and the announcement came with an $18M seed round from Mayo Clinic and Eli Lilly. For anyone building FDA cleared clinical AI, this is the most consequential regulatory signal since the FDA AI/ML action plan.
It matters because a clearly scoped, clinician-supervised, protocol-bounded LLM can clear through 510(k). The pathway is real. It’s also narrow, and most LLM products won’t travel it.
Plenty of people are still arguing whether AI should be regulated at all. If you’re shipping a clinical product, three questions decide your path: does my product need clearance, can it get clearance, and what does clearance require (intended use, predicate, design controls, clinical evidence, post-market surveillance)?
That last question is the build. The first one is what to answer today: which lane are you actually in?
Can a patient-facing clinical LLM get FDA 510(k) clearance, and what does it take?
Yes, UpDoc’s K253281 clearance proved a clinician-supervised, protocol-bounded, patient-facing LLM can clear via 510(k), scoped as a drug-dose calculator against the d-Nav predicate. Clearing one is a five-part build: a predicate, a narrow intended use, design controls under the QMSR, clinical and safety evidence, and a Predetermined Change Control Plan. Most clinical AI never needs clearance, though; the first move is the four-criterion CDS exemption test, where patient-facing output and agentic execution are the two lines that make you a device.
Key Takeaways:
- The 510(k) pathway for patient-facing clinical LLMs is real, and it’s narrow. UpDoc cleared a protocol-bounded insulin tool against a non-LLM predicate, so the precedent reaches insulin and bounded instruction delivery. General agentic clinical AI still needs its own predicate or a different pathway.
- Start with the CDS exemption test, because most clinical AI isn’t a device. The four Cures Act criteria, read more permissively under the January 2026 guidance, decide your lane. Patient-facing direct action and agentic execution are the two lines that put you in device territory, however you frame the physician oversight.
- In the device lane, a rigorous regulatory analysis is the real barrier to entry. The build is concrete: intended use, predicate, design controls under the QMSR, clinical evidence, a PCCP, post-market surveillance. UpDoc’s $18M funded the whole company, and the documented analysis of your intended use is the part that’s hard to copy.
Table of Contents
- What the UpDoc clearance actually establishes (and what it doesn’t)
- The FDA regulatory framework for LLM clinical AI: start with the CDS exemption test
- The 510(k) vs. CDS exemption decision matrix
- The 510(k) pathway for LLM-based SaMD: what it actually requires
- What the $18M seed round tells you, and what it doesn’t
- Post-market surveillance: the obligation that follows clearance
- What the UpDoc clearance means for your build, by product category
- Why Topflight Apps for regulated clinical AI development
What the UpDoc clearance actually establishes (and what it doesn’t)
A patient-facing LLM that delivers clinical instructions within physician-defined parameters can clear FDA review through 510(k). That’s what UpDoc confirmed, and it’s the first LLM medical device FDA clearance to pull it off, cleared as Software as a Medical Device (SaMD). The precedent is concrete. The edges are narrow enough that most teams will misread them, so start with what FDA actually cleared.
What the clearance establishes:
Regulatory identity
FDA slotted UpDoc into an existing drug-dose-calculator code (product code NDC, 21 CFR 868.1890), reviewed by the In Vitro Diagnostics office. There’s no new “AI device” category here. The calculator is the regulatory identity; the model is the interface.
Architecture
The cleared system runs as three modules, a provider portal, a patient app, and a cloud app. The cloud app is where it splits a Conversation Service (the UpDoc Agent, a patient-facing AI agent) from a Clinical Service that computes insulin instructions inside the HCP-set parameters. Physician oversight is structural here, baked into where the math happens, so the model can’t push a dose outside the plan.
Indication
Medication management for adults 18 and older with type 2 diabetes, delivering insulin instructions from an HCP-specified treatment plan. Prescription-only.
Predicate
UpDoc cleared by substantial equivalence to d-Nav (K181916), a patient-facing autonomous insulin titrator cleared back in 2019. Patient-facing autonomous dosing was already on the books; what UpDoc added was the large language model interface. So the precedent reaches insulin and protocol-bounded instruction delivery, and general agentic clinical AI sits outside it.
Two cautions keep this from becoming a template you can copy. Clearing the device and building a HIPAA-compliant app are different regimes; FDA sign-off says nothing about your BAA chain or PHI data flows. And the precedent itself doesn’t stretch as far as the headlines suggest:
- Not every LLM product can clear now. This took real regulatory work, and most LLM tools won’t have a predicate this clean.
- The CDS exemption still covers a lot of clinical software that never needs clearance at all. The next section is how you test for that.
- The pathway isn’t simple or fast. A 510(k) on a strong predicate is still a full design-controls and evidence build.
- There’s no cost or timeline benchmark to take from this. Don’t quote UpDoc’s path to your board as a number; the work scales with your intended use.
- The cleared indication stops at insulin instructions within an HCP plan. It says nothing about writing back to the EHR, so don’t assume EHR write-back rides on this predicate. Scope it into your own intended use.
So the honest read: at the regulatory level, UpDoc is a dosing calculator, and the LLM is how patients talk to it. The architecture pattern is worth studying. Whether your product clears the same way is a separate question, and you still have to answer it.
The FDA regulatory framework for LLM clinical AI: start with the CDS exemption test
The first move in clinical AI FDA regulation is the exemption test. Before clearance is even a question, you check whether your software is a device at all, and plenty of clinical AI isn’t. This is where AI in healthcare compliance starts for a clinical product. Teams get this wrong in two directions: some assume they’re exempt without running the analysis, others assume they need clearance without scoping their intended use. Both are expensive.
That carve-out is the clinical decision support (CDS) exemption, and it runs on a four-part statutory test. Section 520(o)(1)(E) defines software that’s exempt when:
- It doesn’t acquire, process, or analyze a medical image, a signal from an in-vitro diagnostic device, or a pattern or signal from a signal-acquisition system.
- It’s meant to display, analyze, or print medical information about a patient, or other medical information like peer-reviewed studies and clinical guidelines.
- It’s meant to give a healthcare provider recommendations about preventing, diagnosing, or treating a condition.
- It lets that provider independently review the basis for the recommendation, so they’re not relying primarily on the software to make the call.
Fail any one of these and your software is a device. From there, SaMD risk classification sets the pathway and how much evidence you’ll owe.
Four things tend to break the test for LLM products. Patient-facing output is the common one: the moment recommendations go to a patient or caregiver instead of a clinician, Criterion 3 fails, because the exemption only covers HCP-facing software. Black-box reasoning a clinician can’t inspect fails Criterion 4. A continuous signal like a CGM feed trips Criterion 1, since that’s a pattern from a signal-acquisition system. And agentic AI that acts on its own recommendation fails Criterion 4 from the other side, because nobody’s independently reviewing anything before it executes.
A clinician-in-the-loop design is how you hold onto Criterion 4. It only works if the clinician can see the reasoning and override it; a rubber-stamp doesn’t satisfy “independent review.”
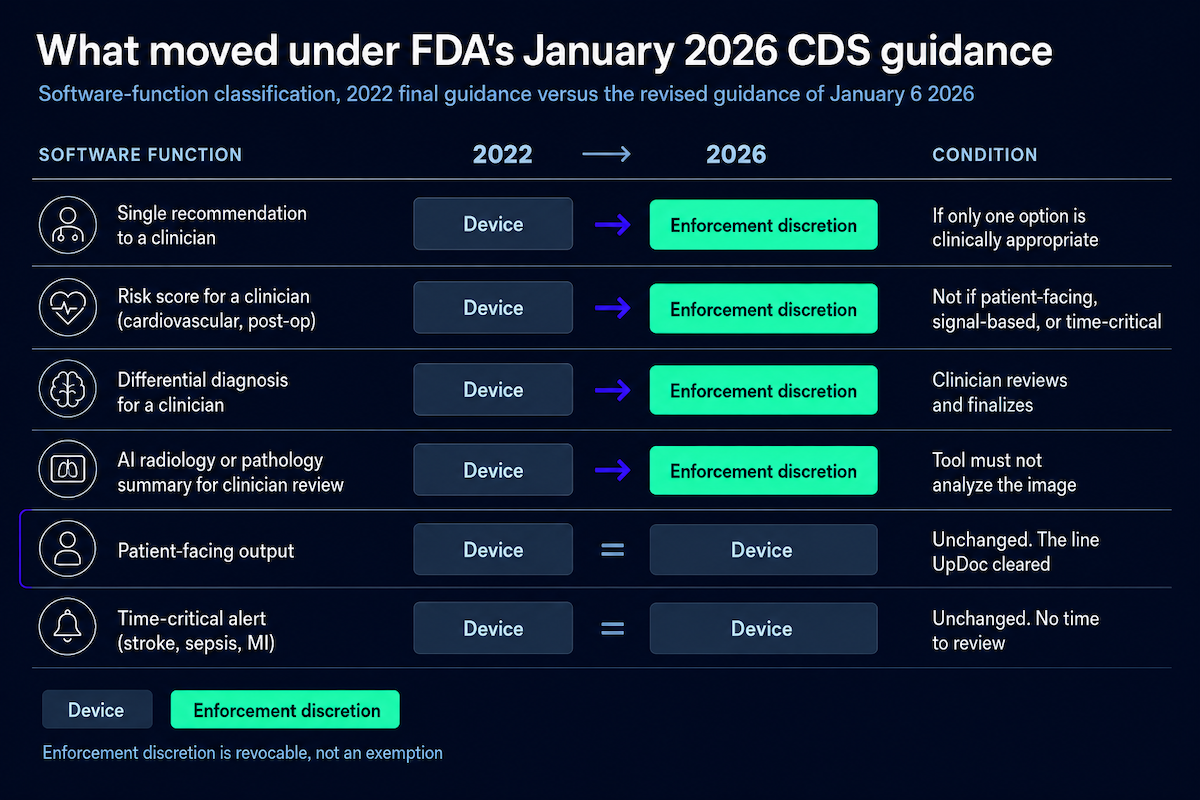
The 21st Century Cures Act CDS exemption itself didn’t change in 2026. The four criteria are word-for-word what they were. What changed is how FDA reads them, and the January 6, 2026 guidance reads them more permissively.
A single-recommendation output can now get enforcement discretion, but only when one option is genuinely the clinically appropriate one. Risk scores and differential diagnoses aimed at an HCP, which the 2022 guidance treated as automatic devices, now fall under enforcement discretion too, and the new examples reach LLM summarization.
That sounds like a lot of room. Read the fine print: enforcement discretion is FDA choosing not to enforce, for now, and it can pull that back per product or per category. A device under enforcement discretion is still a device. And patient-facing software is still a device.
Builder Alert: UpDoc fails this test on two criteria, which is exactly why it needed clearance. Its insulin instructions go straight to patients, so Criterion 3 is out. It ingests CGM data, a continuous signal, so Criterion 1 is out too. Patient-facing clinical instruction delivery lands outside the exemption no matter how you structure the physician oversight around it.
The 510(k) vs. CDS exemption decision matrix
Your regulatory lane comes down to a few concrete characteristics: who acts on the output, whether the software executes anything, and whether it touches diagnostic signals. What the product is called has nothing to do with it. The table maps each one to where it tends to land under the 2026 guidance.
| Product characteristic | CDS exempt? | Device (510(k))? | Notes |
|---|---|---|---|
| Clinician reviews AI output | Likely | No, if all 4 met | Criterion 4 is the one teams miss |
| Patient acts on the output | No | Yes | Fails Criterion 3; this is UpDoc |
| AI recommends, clinician decides | Maybe | Depends | Exempt only if the basis is reviewable (Criterion 4) |
| AI gives a single recommendation | Enforcement discretion | Depends | Only when one option is clinically appropriate |
| AI executes tasks (orders, dose changes) | No | Yes | Fails Criterion 4, no independent review |
| AI processes diagnostic or monitoring signals (IVD, continuous CGM) | No | Yes | Fails Criterion 1 |
| AI scores clinical risk for an HCP | Enforcement discretion (2026) | Conditional | Device if patient-facing or black-box |
| AI generates documentation only (ambient scribe) | Likely | Usually no | If a physician reviews before the note is filed |
| AI coordinates between-visit care | Depends | Yes, if it auto-acts | UpDoc’s lane |
Land anywhere in the device column and you’re headed for AI SaMD 510(k) clearance. A few rows carry most of the weight. Patient acts on the output and you’re a device, no matter how the oversight is framed. AI that executes, placing the order or changing the dose, is a device, because there’s no room for independent review.
Anything processing diagnostic or monitoring signals, an IVD readout or a continuous CGM stream, is a device under Criterion 1. The risk-score row is the one that moved: an HCP-facing risk score used to be an automatic device, and under the 2026 guidance it now gets enforcement discretion. Ambient documentation usually stays exempt, as long as a physician reviews the draft before it reaches the record.

UpDoc itself sits in two of these rows, patient-acting-on-output and between-visit care coordination. A single clinical workflow rarely collapses to one function, so run each software function through the table on its own.
EHR-integrated AI is the usual culprit: a scribe that also surfaces a risk flag is two functions, and each classifies separately. And keep the two right-hand outcomes straight. An exemption means you’re not a device at all. Enforcement discretion is different: you’re a device, and FDA is just choosing not to enforce against you today.
The 510(k) pathway for LLM-based SaMD: what it actually requires
LLM SaMD development through 510(k) is five linked deliverables: a predicate, an intended use, design controls, clinical evidence, and a change-control plan. Each one feeds the next. This is the health AI FDA clearance build, and UpDoc shows how each piece actually got done.
Predicate strategy is the hardest part
The whole 510(k) clearance rests on substantial equivalence, so you need a predicate device: something already cleared that does the same clinical job, plus an argument that your technology does it equivalently. UpDoc pointed at d-Nav (K181916), a cleared insulin titrator. Same intended use, patient-facing insulin titration inside a provider-set plan. The one thing UpDoc had to argue as new was the LLM voice-and-text interface.
That’s why it cleared in 85 days. d-Nav was already patient-facing and already evidence-backed: a Lancet RCT (Bergenstal 2019, n=181) where HbA1c dropped 1.0% against 0.3% in the control arm at six months (p<0.0001). With a predicate that strong, the substantial-equivalence argument is short.
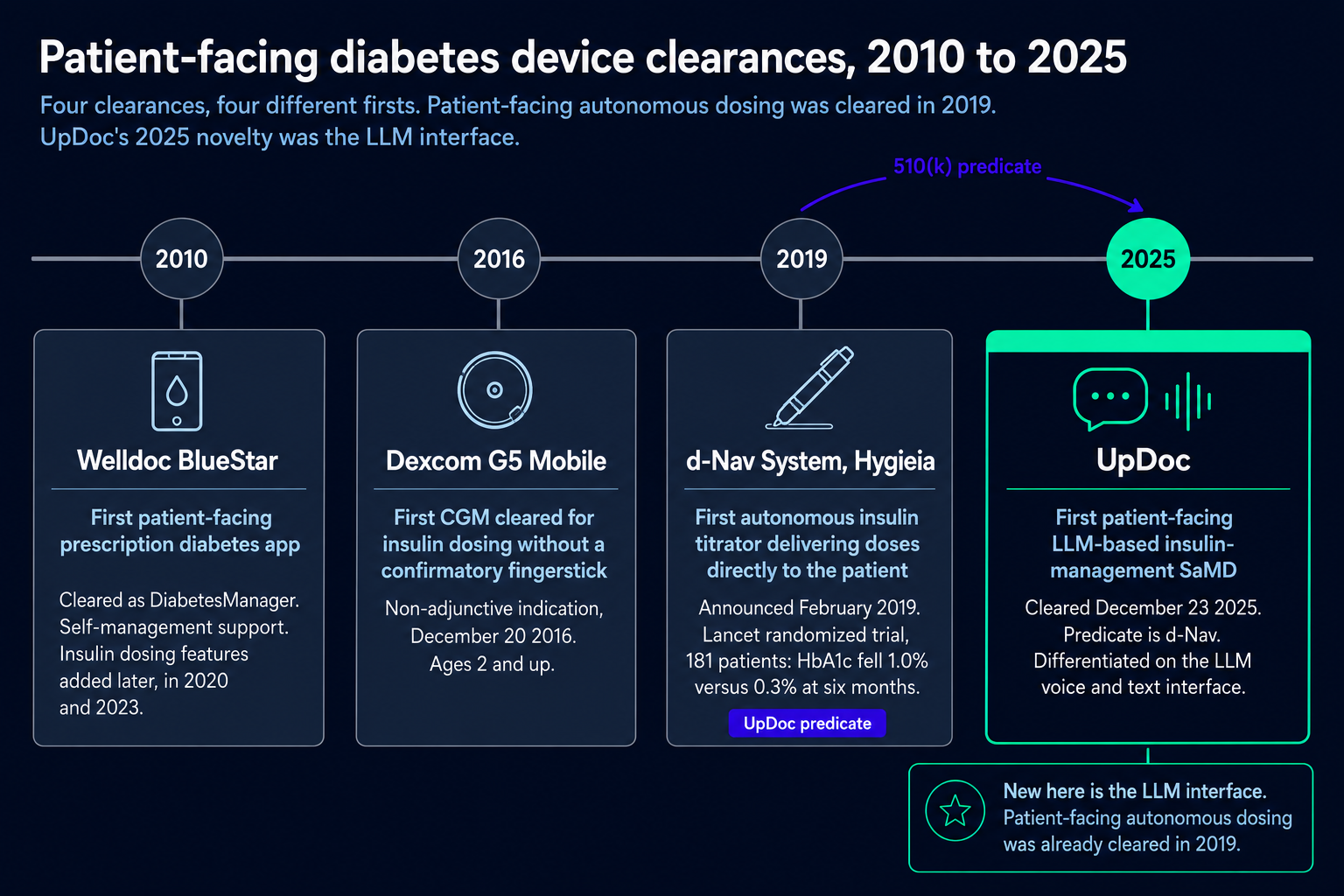
Post-UpDoc, the math changes. K253281 is now a predicate, but a narrow one: insulin, protocol-bounded patient instruction. General agentic clinical AI doesn’t have a clean predicate here, so you’re looking at a different cleared device or a De Novo. If your intended use lines up with an existing device but your technology doesn’t, a split predicate (one for the use, one for the technology) is worth raising with regulatory counsel.
Intended use is the single most important document you’ll write
Your intended use statement decides everything downstream: your classification, which predicate fits, and what evidence you owe. So write it narrow, and write it concrete. Three things have to be unambiguous:
- the user (a patient, or a clinician)
- the clinical task (what the software does, in plain clinical terms)
- the boundary (the parameters it operates inside)
Patient-facing intended use draws more scrutiny, and it’s still clearable. UpDoc’s indications for use are the model to copy: medication management for patients 18 and older with type 2 diabetes, delivering insulin treatment-plan instructions from an HCP-specified plan. And the phrasing has to be functional. “AI-powered clinical assistance” gives FDA nothing to work with. “Medication-adjustment facilitation within physician-approved parameters” gives them a device they can classify.
Design controls under QMSR
Design controls are a documented framework: design inputs, design outputs, verification, validation, and design transfer, each producing a required artifact. Good engineering habits help you generate them; the artifacts are what FDA actually reviews. For an LLM, the interesting part is what goes into each:
- design inputs: accuracy, latency, a hallucination-rate tolerance, and a clinician-override mechanism
- design validation: data from the real clinical environment, beyond what a retrospective record review can show
- design transfer: model and prompt versioning, plus the PCCP that governs how they change
PHI data flows belong in your design inputs too. If you’re routing patient data through a third-party model, ChatGPT HIPAA compliance is exactly the kind of question to settle at design time, before it surfaces in an audit.
All of this runs under the QMSR, effective February 2, 2026, which folds ISO 13485:2016 into Part 820 by reference. FDA hasn’t published 510(k)-specific guidance on presenting your QMS in a submission yet, only a PMA/HDE draft from October 2025. The QMSR governs your quality system regardless.
Clinical evidence
Clinical validation of an LLM is a heavier lift than it is for rule-based software. UpDoc’s pathway didn’t require a trial at all, since substantial equivalence carried it, and UpDoc ran one anyway. The numbers are company-reported and not peer-reviewed, so attribute them: 81% of AI-managed patients hit glycemic control at 8 weeks against 25% on traditional care, with 60% greater medication adherence.
Whether or not you run a trial, expect to show evidence on:
- clinical performance on the actual intended task
- hallucination-rate and safety data
- AI bias and fairness across age, sex, race, and ethnicity
- clinician-override effectiveness (does the human actually catch the bad output)

On cost, a software-only submission moves the spend from physical testing to software validation and cybersecurity documentation. Only about 10% of 510(k)s need a human clinical study at all. Well-prepared, with a predicate as clean as d-Nav, the all-in lands well under $200K. The FY2026 user fee is $26,067, or $6,517 with small-business status, which UpDoc qualified for.
The Predetermined Change Control Plan (PCCP)
The PCCP is how you update the model without filing a new 510(k). UpDoc’s cleared as part of K253281 under §515C, so the plan to keep improving the model got reviewed up front.
A good LLM PCCP spells out the changes you’ll actually make, each with its own validation protocol: new model versions, prompt-template edits, retrieval-augmented generation (RAG) knowledge-base updates, and LLM fine-tuning. The catch is timing. A PCCP has to be approved in the original 510(k), and you can’t file one retroactively. A model you intend to keep improving needs its change plan written before you clear.
What the $18M seed round tells you, and what it doesn’t
Start with what the round does tell you. $18M, from Mayo Clinic and Eli Lilly, is institutional money backing regulated clinical AI from day one. Two of the most conservative names in healthcare wrote that check before the product had scaled. That’s the signal: serious capital now treats the regulatory build as the strategy, funded from the start, in a category that used to scare generalist investors off.
What it doesn’t tell you is the price of clearance. People see a big raise sitting next to an FDA clearance and assume the two are linked. They’re not. The $18M is not what a 510(k) costs. UpDoc cleared in 85 days, qualified for the small-business fee, and a well-prepared software submission with a predicate this strong lands well under $200K. The round funds the whole company: the team, the product, and the competitive position a clearance creates. That last part is the real prize, a regulatory moat most competitors won’t have.
For an investor or an exec doing diligence, the useful shift is in the question. The old reflex was to check for AI medical software FDA approval as a yes-or-no box. Most products don’t have it, and plenty don’t need it. The sharper question now: has this team run a real regulatory analysis of its intended use, and is it in the right lane? A documented answer to that is the lower-risk bet.
UpDoc’s CEO, Sharif Vakili, MD, has said clinical AI “should be held to the highest standard.” The capital is betting he’s right.
Post-market surveillance: the obligation that follows clearance
Clearance starts a post-market surveillance obligation that runs for the life of the product. The hardest part of it, for an LLM, is one you have to solve before you ship: what counts as a reportable malfunction when the failure mode is a hallucination?
Four obligations kick in the day you clear:
- Adverse event reporting under MDR (21 CFR Part 803): death or serious injury or a reportable malfunction goes to FDA within 30 calendar days, and within 5 working days when remedial action is needed to head off an unreasonable risk of substantial harm.
- Ongoing QMSR duties under 21 CFR Part 820: CAPA, complaint handling, and document control don’t stop at clearance; they run continuously.
- Post-market performance monitoring: FDA expects real-world data measured against the validation data you submitted in the 510(k).
- PCCP thresholds: if performance degrades below the level you validated, that can trigger a notification under the PCCP you cleared.
The LLM-specific work is in the definitions. “Reportable malfunction” is obvious for a pump that stops pumping; for an LLM you have to define it yourself, a hallucination that drives a harmful decision, or an oversight step that silently fails. That definition is a design and QMS requirement written before launch, because you can’t report against a malfunction you never specified. Complaint handling is its own puzzle: when users talk to the product in plain language, you need a way to tell ordinary dissatisfaction apart from a real safety signal, and to push that signal into CAPA fast. And real-world monitoring means watching a moving target, because model or data drift can pull live performance below your validated baseline with no code change at all.
Enforcement here is real. By one analysis of FY2025 device warning letters, 38 of 44 cited Part 820. FDA also retired the old QSIT inspection model on February 2, 2026 and moved to a new compliance program. Post-market, the quality system is where FDA tends to find you, so build your patient safety monitoring to survive an inspection from the start.
What the UpDoc clearance means for your build, by product category
The clearance doesn’t move every category equally. What the first FDA cleared LLM healthcare product means for your build depends on what your software actually does, and for two categories it means re-testing an exemption you’ve been assuming.
Ambient AI scribes
Standard ambient scribes, the ones that draft a note for a physician to review before it hits the record, generally stay exempt. AI clinical documentation that stops at transcription and drafting is HCP-facing with a human in the loop. Nuance DAX and Abridge live here. The 2026 guidance adds a wrinkle worth knowing: an AI-drafted summary that includes a diagnostic recommendation for the physician to review and finalize now falls under enforcement discretion, where the 2022 guidance would have called it a device. Enforcement discretion is revocable, though, so if you’re adding a clinical-inference layer on top of the scribe, re-test your lane before you ship it. Ambient AI scribe development sits right on this line.
Care management and between-visit engagement
This is the category the clearance hits hardest: between-visit care and care management. UpDoc is now the live precedent, patient-facing between-visit insulin adjustment, inside an HCP plan, cleared as SaMD. If you’re building a clinical medication adherence platform, patient-facing medication adjustment AI, or any care team coordination AI that takes an automated action between visits, the assumed CDS exemption a lot of these products ride on just got weaker. Run a formal regulatory analysis of your intended use before your next raise or enterprise sale, because that’s when a sophisticated counterparty will ask.
Clinician-facing CDS
Clinician-facing CDS is the most defensible exemption there is. A clinician reads the reasoning and makes the call, and nothing executes on its own. The UpDoc clearance is reassuring here: it marks the edge of the exemption, patient-facing output and agentic execution, and leaves everything inside that edge exactly where it was.
Diagnostic AI
Diagnostic AI is already SaMD, and the 2026 guidance didn’t change that. Software that analyzes an image or a signal to produce a finding trips Criterion 1. What UpDoc adds is a different signal: it shows the 510(k) pathway works for AI that’s woven into clinical workflow, which may lower the perceived barrier for diagnostic teams who’ve been treating clearance as a wall. The AI in the EHR question sits next to this one for anyone integrating diagnostic output into the record.
Why Topflight Apps for regulated clinical AI development
You’ve just seen the whole regulated-AI stack mapped out: exemption test, predicate, design controls, clinical evidence, PCCP, post-market surveillance. That’s the work we do at Topflight Apps. Plenty of regulatory consultancies can’t ship code, and plenty of dev shops have never seen a 510(k). We’re the shop that does both, engineering and regulatory together, with a decade of HIPAA-bounded healthcare builds and dozens of EHR integrations behind us. We map where you sit, then build what clearance needs:
- Regulatory readiness assessment: we map your intended use to the CDS exemption and the right SaMD classification.
- Design-controls implementation under the QMSR.
- Clinical validation planning: LLM accuracy, hallucination rate, bias and fairness, and clinician-oversight effectiveness.
- PCCP drafting for how you’ll actually update the model: versions, prompts, RAG, and fine-tuning.
- EHR integration on FHIR R4, SMART on FHIR, and CDS Hooks, including Epic integration.
- HIPAA for AI systems: HIPAA-compliant software development for LLMs, covering the business associate agreement (BAA) chain, PHI data-flow documentation, subprocessor mapping, and audit logging.
If regulated clinical AI development is on your roadmap, talk to us before you write the intended-use statement. That’s the document everything else hangs on, and it’s the cheapest place to get the strategy right.
Frequently Asked Questions
What is the difference between the CDS software exemption and FDA device classification?
Software meeting all four Cures Act criteria is exempt: HCP-facing, reviewable basis, no diagnostic-signal processing, no patient-facing direct action. Fail any one and it’s a device.
Can a patient-facing LLM receive FDA 510(k) clearance?
UpDoc’s December 2025 clearance (K253281) covered patient-facing insulin instructions within an HCP plan, using the non-LLM d-Nav predicate. The pathway required no clinical trial.
What is a Predetermined Change Control Plan (PCCP), and why does it matter for LLM products?
It’s the FDA-approved plan that lets you update the model, versions, prompts, RAG, fine-tuning, without a new 510(k). It must be in the original submission; UpDoc’s cleared with K253281.
How much does a 510(k) clearance cost for an AI medical device?
Typically $50K to $250K all-in, plus the FY2026 user fee of $26,067 ($6,517 for small businesses). For software-only, with a strong predicate, well-prepared submissions land under $200K.
What clinical evidence does FDA require for an LLM-based SaMD submission?
Performance on the intended task, hallucination and safety data, bias and fairness across subgroups, and clinician-override effectiveness. Trials aren’t always required, UpDoc’s weren’t, but expect to show evidence.